 -
Volume 150,
Issue 6,
2004
-
Volume 150,
Issue 6,
2004

Volume 150, Issue 6, 2004
- BIOCHEMISTRY AND MOLECULAR BIOLOGY
-
-
-
The Mycobacterium tuberculosis cysD and cysNC genes form a stress-induced operon that encodes a tri-functional sulfate-activating complex
More LessSulfur metabolism has been implicated in the virulence, antibiotic resistance and anti-oxidant defence of Mycobacterium tuberculosis. Despite its human disease relevance, sulfur metabolism in mycobacteria has not yet been fully characterized. ATP sulfurylase catalyses the synthesis of activated sulfate (adenosine 5′-phosphosulfate, APS), the first step in the reductive assimilation of sulfate. Expression of the M. tuberculosis cysD gene, predicted to encode the adenylyl-transferase subunit of ATP sulfurylase, is upregulated by the bacilli inside its preferred host, the macrophage. This study demonstrates that cysD and cysNC orthologues exist in M. tuberculosis and constitute an operon whose expression is induced by sulfur limitation and repressed by the presence of cysteine, a major end-product of sulfur assimilation. The cysDNC genes are also induced upon exposure to oxidative stress, suggesting regulation of sulfur assimilation by M. tuberculosis in response to toxic oxidants. To ensure that the cysDNC operon encoded the activities predicted by its primary sequence, and to begin to characterize the products of the operon, they were expressed in Escherichia coli, purified to homogeneity, and tested for their catalytic activities. The CysD and CysNC proteins were shown to form a multifunctional enzyme complex that exhibits the three linked catalytic activities that constitute the sulfate activation pathway.
-
-
-
-
Rhodococcus opacus expresses the xsc gene to utilize taurine as a carbon source or as a nitrogen source but not as a sulfur source
More LessThe Gram-positive bacteria Rhodococcus opacus ISO-5 and Rhodococcus sp. RHA1 utilized taurine (2-aminoethanesulfonate) as the sole source of carbon or of nitrogen or of sulfur for growth. Different gene clusters and enzymes were active under these different metabolic situations. Under carbon- or nitrogen-limited conditions three enzymes were induced, though to different levels: taurine-pyruvate aminotransferase (Tpa), alanine dehydrogenase (Ald) and sulfoacetaldehyde acetyltransferase (Xsc). The specific activities of these enzymes in R. opacus ISO-5 were sufficient to explain the growth rates under the different conditions. These three enzymes were purified and characterized, and the nature of each reaction was confirmed. Analyses of the genome of Rhodococcus sp. RHA1 revealed a gene cluster, tauR-ald-tpa, putatively encoding regulation and oxidation of taurine, located 20 kbp from the xsc gene and separate from two candidate phosphotransacetylase (pta) genes, as well as many candidate ABC transporters (tauBC). PCR primers allowed the amplification and sequencing of the tauR-ald-tpa gene cluster and the xsc gene in R. opacus ISO-5. The N-terminal sequences of the three tested proteins matched the derived amino acid sequences of the corresponding genes. The sequences of the four genes found in each Rhodococcus strain shared high degrees of identity (>95 % identical positions). RT-PCR studies proved transcription of the xsc gene when taurine was the source of carbon or of nitrogen. Under sulfur-limited conditions no xsc mRNA was generated and no Xsc was detected. Taurine dioxygenase (TauD), the enzyme catalysing the anticipated desulfonative reaction when taurine sulfur is assimilated, was presumed to be present because oxygen-dependent taurine disappearance was demonstrated with taurine-grown cells only. A putative tauD gene (with three other candidates) was detected in strain ISO-5. Regulation of the different forms of metabolism of taurine remains to be elucidated.
-
-
-
Characterization of the Streptomyces lavendulae IMRU 3455 linear plasmid pSLV45
More LessStreptomyces lavendulae IMRU 3455 contains two large linear plasmids designated pSLV45 (45 kb) and pSLV195 (195 kb). A cosmid, pSPRX604, containing 42 kb from pSLV45 was cloned and sequenced. pSLV45 was tagged with a hygromycin-resistance marker by homologous recombination to generate the derivatives pSLV45.680 and pSLV45.681. An apramycin-resistance marker was introduced into S. lavendulae IMRU 467 using the pSPR910 integration vector to yield the recipient strain SPW910. The self-transmissible nature of pSLV45 was determined by transfer of pSLV45.680 and pSLV45.681 from the donor strains SPW680 and SPW681 into the recipient strain SPW910. Southern analysis indicated the presence of hygromycin- and pSLV45-hybridizing sequences within SPW910 exconjugants. PFGE analysis confirmed pSLV45.680 and pSLV45.681 were transferred intact and formed freely replicating linear plasmids. Sequence analysis of pSPRX604 revealed genes predicted to be involved in plasmid transfer, partitioning and regulation. The transfer of the linear plasmid pSLV45 from S. lavendulae IMRU 3455 into S. lavendulae IMRU 467 may allow the development of pSLV45 as an actinomycete-to-actinomycete conjugative shuttle vector.
-
-
-
DNA methyltransferases from Neisseria meningitidis and Neisseria gonorrhoeae FA1090 associated with mismatch nicking endonucleases
More LessThe genes encoding the DNA methyltransferases M.NmeDI and M.NmeAI from Neisseria meningitidis associated with the genes encoding putative Vsr endonucleases were overexpressed in Escherichia coli. The enzymes were purified to apparent homogeneity on Ni-NTA agarose columns, yielding proteins of 49±1 kDa and 39·6±1 kDa, respectively, under denaturing conditions. M.NmeDI recognizes the degenerate sequence 5′-RCCGGB-3′. It methylates the first 5′ cytosine residue on both strands within the core sequence CCGG. The enzyme shows higher affinity with the hemimethylated degenerate sequence than with the unmethylated degenerate sequence. Comparison of the amino acid sequence of the target-recognizing domain of M.NmeDI with the closest neighbours recognizing the sequence 5′-RCCGGY-3′ showed the presence of the homologous domain and an additional domain that may be responsible for recognizing the degenerate sequence. M.NmeAI recognizes the sequence 5′-CCGG-3′ and methylates the second 5′ cytosine residue on both DNA strands. In Neisseria gonorrhoeae strain FA1090 the homologues of these ORFs are truncated due to a variety of mutations.
-
-
-
luxS mutants of Serratia defective in autoinducer-2-dependent ‘quorum sensing’ show strain-dependent impacts on virulence and production of carbapenem and prodigiosin
More LessThe enzyme LuxS is responsible for the production of autoinducer-2 (AI-2), a molecule that has been implicated in quorum sensing in many bacterial species. This study investigated whether there is a luxS-dependent signalling system in the Gram-negative bacteria Serratia spp. Serratia marcescens is a broad-host-range pathogen and an important cause of nosocomial infections. Production of AI-2 activity was detected in S. marcescens ATCC 274 and Serratia ATCC 39006 and their luxS genes were sequenced. luxS mutants were constructed in these strains and were analysed to determine which phenotypes are regulated by luxS and therefore, potentially, by AI-2. The phenotypes of the luxS mutants included decreased carbapenem antibiotic production in Serratia ATCC 39006 and decreased prodigiosin and secreted haemolysin production in S. marcescens ATCC 274. The luxS mutant of S. marcescens ATCC 274 was also found to exhibit modestly reduced virulence in a Caenorhabditis elegans model. Finally, it was shown that the culture supernatant of a wild-type strain contains a signal, presumably AI-2, capable of complementing the prodigiosin defect of the luxS mutant of another strain, even when substantially diluted. It is concluded that luxS modulates virulence and antibiotic production in Serratia, in a strain-dependent manner, and that, for at least one phenotype, this regulation is via extracellular signalling.
-
-
-
The role of dor gene products in controlling the P2 promoter of the cytochrome c 2 gene, cycA, in Rhodobacter sphaeroides
More LessThis study explores the regulatory networks controlling anaerobic energy production by the facultative phototroph Rhodobacter sphaeroides. The specific aim was to determine why activity of the P2 promoter for the gene (cycA) encoding the essential photosynthetic electron carrier, cytochrome c 2, is decreased when the alternative electron acceptor DMSO is added to photosynthetically grown cells. The presence of DMSO is believed to activate the DorR response regulator, which controls expression of proteins required to reduce DMSO. A DorR− strain showed no change in cycA P2 promoter activity when DMSO was added to photosynthetic cells, indicating that DorR was required for the decreased expression in wild-type cells. To test if DorR acted directly at this promoter to change gene expression, recombinant DorR was purified and studied in vitro. Preparations of DorR that were active at other target promoters showed no detectable interaction with cycA P2, suggesting that this protein is not a direct regulator of this promoter. We also found that cycA P2 activity in a DorA− strain was not decreased by the addition of DMSO to photosynthetic cells. A model is presented to explain why the presence of a functional DMSO reductase (DorA) is required for DMSO to decrease cycA P2 expression under photosynthetic conditions.
-
-
-
FpvB, an alternative type I ferripyoverdine receptor of Pseudomonas aeruginosa
More LessUnder conditions of iron limitation, Pseudomonas aeruginosa secretes a high-affinity siderophore pyoverdine to scavenge Fe(III) in the extracellular environment and shuttle it into the cell. Uptake of the pyoverdine–Fe(III) complex is mediated by a specific outer-membrane receptor protein, FpvA (ferripyoverdine receptor). Three P. aeruginosa siderovars can be distinguished, each producing a different pyoverdine (type I–III) and a cognate FpvA receptor. Growth of an fpvA mutant of P. aeruginosa PAO1 (type I) under iron-limiting conditions can still be stimulated by its cognate pyoverdine, suggesting the presence of an alternative uptake route for type I ferripyoverdine. In silico analysis of the PAO1 genome revealed that the product of gene PA4168 has a high similarity with FpvA. Inactivation of PA4168 (termed fpvB) in an fpvA mutant totally abolished the capacity to utilize type I pyoverdine. The expression of fpvB is induced by iron limitation in Casamino acids (CAA) and in M9-glucose medium, but, unlike fpvA, not in a complex deferrated medium containing glycerol as carbon source. The fpvB gene was also detected in other P. aeruginosa isolates, including strains producing type II and type III pyoverdines. Inactivation of the fpvB homologues in these strains impaired their capacity to utilize type I ferripyoverdine as a source of iron. Accordingly, introduction of fpvB in trans restored the capacity to utilize type I ferripyoverdine.
-
-
-
A novel glucan-binding protein with lipase activity from the oral pathogen Streptococcus mutans
More LessStreptococcus mutans produces extracellular glucosyltransferases (GTFs) that synthesize glucans from sucrose. These glucans are important in determining the permeability properties and adhesiveness of dental plaque. GTFs and the GbpA glucan-binding protein are characterized by a binding domain containing a series of 33-amino-acid repeats, called ‘A’ repeats. The S. mutans genome sequence was searched for ORFs containing ‘A’ repeats, and one novel gene, gbpD, which appears to be unique to the mutans group of streptococci, was identified. The GbpD sequence revealed the presence of three ‘A’ repeats, in the middle of the protein, and a novel glucan-binding assay showed that GbpD binds to dextran with a K D of 2–3 nM. Construction of truncated derivatives of GbpD confirmed that the ‘A’ repeat region was essential for binding. Furthermore, a gbpD knockout mutant was modified in the extent of aggregation induced by polymers derived from sucrose. The N-terminus of GbpD has a signal sequence, followed by a region with no homologues in the public databases, while the C-terminus has homology to the α/β hydrolase family (including lipases and carboxylesterases). GbpD contains the two regions typical of these enzymes: a GxSxG active site ‘lipase box’ and an ‘oxyanion hole’. GbpD released free fatty acids (FFAs) from a range of triglycerides in the presence of calcium, indicating a lipase activity. The glucan binding/lipase bifunctionality suggested the natural substrate for the enzyme may be a surface macromolecule consisting of carbohydrate linked to lipid. The gbpD mutant was less hydrophobic than wild-type and pure recombinant GbpD reduced the hydrophobicity of S. mutans and another plaque bacterium, Streptococcus sanguinis. GbpD bound to and released FFA from lipoteichoic acid (LTA) of S. sanguinis, but had no effect on LTA from S. mutans. These results raise the intriguing possibility that GbpD may be involved in direct interspecies competition within the plaque biofilm.
-
-
-
Saccharomyces kluyveri FAD3 encodes an ω3 fatty acid desaturase
More LessFungi, like plants, are capable of producing the 18-carbon polyunsaturated fatty acids linoleic acid and α-linolenic acid. These fatty acids are synthesized by catalytic reactions of Δ12 and ω3 fatty acid desaturases. This paper describes the first cloning and functional characterization of a yeast ω3 fatty acid desaturase gene. The deduced protein encoded by the Saccharomyces kluyveri FAD3 gene (Sk-FAD3) consists of 419 amino acids, and shows 30–60 % identity with Δ12 fatty acid desaturases of several eukaryotic organisms and 29–31 % identity with ω3 fatty acid desaturases of animals and plants. During Sk-FAD3 expression in Saccharomyces cerevisiae, α-linolenic acid accumulated only when linoleic acid was added to the culture medium. The disruption of Sk-FAD3 led to the disappearance of α-linolenic acid in S. kluyveri. These findings suggest that Sk-FAD3 is the only ω3 fatty acid desaturase gene in this yeast. Furthermore, transcriptional expression of Sk-FAD3 appears to be regulated by low-temperature stress in a manner different from the other fatty acid desaturase genes in S. kluyveri.
-
-
-
AgmR controls transcription of a regulon with several operons essential for ethanol oxidation in Pseudomonas aeruginosa ATCC 17933
More LessThe response regulator AgmR was identified to be involved in the regulation of the quinoprotein ethanol oxidation system of Pseudomonas aeruginosa ATCC 17933. Interruption of the agmR gene by insertion of a kanamycin-resistance cassette resulted in mutant NG3, unable to grow on ethanol. After complementation with the intact agmR gene, growth on ethanol was restored. Transcriptional lacZ fusions were used to identify four operons which are regulated by the AgmR protein: the exaA operon encodes the pyrroloquinoline quinone (PQQ)-dependent ethanol dehydrogenase, the exaBC operon encodes a soluble cytochrome c 550 and an aldehyde dehydrogenase, the pqqABCDE operon carries the PQQ biosynthetic genes, and operon exaDE encodes a two-component regulatory system which controls transcription of the exaA operon. Transcription of exaA was restored by transformation of NG3 with a pUCP20T derivative carrying the exaDE genes under lac-promoter control. These data indicate that the AgmR response regulator and the exaDE two-component regulatory system are organized in a hierarchical manner. Gene PA1977, which appears to form an operon with the agmR gene, was found to be non-essential for growth on ethanol.
-
-
-
Molecular characterization of protein O-mannosyltransferase and its involvement in cell-wall synthesis in Aspergillus nidulans
More LessProtein O-glycosylation is essential for protein modification and plays important roles in eukaryotic cells. O-Mannosylation of proteins occurs in the filamentous fungus Aspergillus. The structure and function of the pmtA gene, encoding protein O-d-mannosyltransferase, which is responsible for the initial O-mannosylation reaction in Aspergillus nidulans, was characterized. Disruption of the pmtA gene resulted in the reduction of in vitro protein O-d-mannosyltransferase activity to 6 % of that of the wild-type strain and led to underglycosylation of an extracellular glucoamylase. The pmtA disruptant exhibited abnormal cell morphology and alteration in carbohydrate composition, particularly reduction in the skeletal polysaccharides in the cell wall. The results indicate that PmtA is required for the formation of a normal cell wall in A. nidulans.
-
-
-
Effects of high hydrostatic pressure on bacterial cytoskeleton FtsZ polymers in vivo and in vitro
More LessSome rod-shaped bacteria, including Escherichia coli, exhibit cell filamentation without septum formation under high-hydrostatic-pressure conditions, indicating that the cell-division process is affected by hydrostatic pressure. The effects of elevated pressure on FtsZ-ring formation in E. coli cells were examined using indirect immunofluorescence microscopy. Elevated pressure of 40 MPa completely inhibited colony formation of E. coli cells under the cultivation conditions used, and the cells exhibited obviously filamentous shapes. In the elongated cells, normal cell-division processes appeared to be inhibited, because no FtsZ rings were observed by indirect immunofluorescent staining. In addition, it was observed that hydrostatic pressure dissociated the E. coli FtsZ polymers in vitro. These results suggest that high hydrostatic pressure directly affects cell survival and morphology through the dissociation of the cytoskeletal frameworks.
-
-
-
The RAM1 gene encoding a protein-farnesyltransferase β-subunit homologue is essential in Cryptococcus neoformans
More LessMany small G proteins require post-translational modification to allow functional association to the cell membrane. This process often involves the enzymic addition of hydrophobic prenyl groups to a conserved cysteine residue near the C-terminus of the protein. The enzymes that catalyse these reactions include protein farnesyltransferase and protein geranylgeranyltransferases. The human fungal pathogen Cryptococcus neoformans requires functional Ras and Rho proteins in order to undergo normal growth and differentiation. Since farnesylation and geranylgeranylation are likely required for the proper function of these small G proteins, we hypothesized that inhibition of these prenylation events would alter the growth and cellular morphogenesis of this fungus. We cloned the RAM1 gene encoding the single protein-farnesyltransferase β-chain homologue in C. neoformans. Using a gene-disruption strategy in a diploid C. neoformans strain, we demonstrated that this gene encodes an essential function, in contrast to the case in Saccharomyces cerevisiae, in which the homologous RAM1 gene is not essential for growth. Pharmacological inhibition of farnesyltransferase activity resulted in dose-dependent cytostasis of C. neoformans, as well as prevention of hyphal differentiation. Simultaneous inhibition of farnesylation and calcineurin signalling results in a synthetic effect on growth. Protein farnesylation is required for the growth and cellular differentiation of C. neoformans and may provide novel targets for antifungal therapy.
-
- BIODIVERSITY AND EVOLUTION
-
-
-
Evolution of compatible replicons of the related IncQ-like plasmids, pTC-F14 and pTF-FC2
More LessTwo closely related but compatible plasmids of the IncQ-2α and IncQ-2β groups, pTF-FC2 and pTC-F14, were discovered in two acidiphilic chemolithotrophic bacteria. Cross-complementation and cross-regulation experiments by the replication proteins were carried out to discover what changes were necessary when the plasmids evolved to produce two incompatibility groups. The requirement of a pTC-F14 oriV for a RepC DNA-binding protein was plasmid specific, whereas the requirement for the RepA helicase and RepB primase was less specific and could be complemented by the IncQ-2α plasmid pTC-FC2, and the IncQ-1β plasmid pIE1108. None of the IncQ-1α plasmid replication proteins could complement the pTC-F14 oriV, and pTC-F14 and RSF1010 were incompatible. This incompatibility was associated with the RepC replication protein and was not due to iteron incompatibility. Replication of pTC-F14 took place from a 5·7 kb transcript that originated upstream of the mobB gene located within the region required for mobilization. A pTC-F14 mobB–lacZ fusion was regulated by the pTC-F14 repB gene product and was plasmid specific, as it was not regulated by the RepB proteins of pTF-FC2 or the IncQ-1α and IncQ-1β plasmids. Plasmid pTC-F14 appears to have evolved independently functioning iterons and a plasmid-specific RepC-binding protein; it also has a major replication transcript that is independently regulated from that of pTF-FC2. However, the RepA and RepB proteins have the ability to function with either replicon.
-
-
-
-
Sequence typing reveals extensive strain diversity of the Lyme borreliosis agents Borrelia burgdorferi in North America and Borrelia afzelii in Europe
More LessThe genetic polymorphism of Borrelia burgdorferi and Borrelia afzelii, two species that cause Lyme borreliosis, was estimated by sequence typing of four loci: the rrs–rrlA intergenic spacer (IGS) and the outer-membrane-protein gene p66 on the chromosome, and the outer-membrane-protein genes ospA and ospC on plasmids. The major sources of DNA for PCR amplification and sequencing were samples of the B. burgdorferi tick vector Ixodes scapularis, collected at a field site in an endemic region of the north-eastern United States, and the B. afzelii vector Ixodes ricinus, collected at a similar site in southern Sweden. The sequences were compared with those of reference strains and skin biopsy isolates, as well as database sequences. For B. burgdorferi, 10–13 alleles for each of the 4 loci, and a total of 9 distinct clonal lineages with linkage of all 4 loci, were found. For B. afzelii, 2 loci, ospC and IGS, were examined, and 11 IGS genotypes, 12 ospC alleles, and a total of 9 linkage groups were identified. The genetic variants of B. burgdorferi and B. afzelii among samples from the field sites accounted for the greater part of the genetic diversity previously reported from larger areas of the north-eastern United States and central and northern Europe. Although ospC alleles of both species had higher nucleotide diversity than other loci, the ospC locus showed evidence of intragenic recombination and was unsuitable for phylogenetic inference. In contrast, there was no detectable recombination at the IGS locus of B. burgdorferi. Moreover, beyond the signature nucleotides that specified 10 IGS genotypes, there were additional nucleotide polymorphisms that defined a total of 24 subtypes. Maximum-likelihood and parsimony cladograms of B. burgdorferi aligned IGS sequences revealed the subtype sequences to be terminal branches of clades, and the existence of at least three monophyletic lineages within B. burgdorferi. It is concluded that B. burgdorferi and B. afzelii have greater genetic diversity than had previously been estimated, and that the IGS locus alone is sufficient for strain typing and phylogenetic studies.
-
-
-
Phylogeny of Mycobacterium avium strains inferred from glycopeptidolipid biosynthesis pathway genes
More LessThe Mycobacterium avium complex (MAC) encompasses two species, M. avium and Mycobacterium intracellulare, which are opportunistic pathogens of humans and animals. The standard method of MAC strain differentiation is serotyping based on a variation in the antigenic glycopeptidolipid (GPL) composition. To elucidate the relationships among M. avium serotypes a phylogenetic analysis of 13 reference and clinical M. avium strains from 8 serotypes was performed using as markers two genomic regions (890 bp of the gtfB gene and 2150 bp spanning the rtfA–mtfC genes) which are associated with the strains' serological properties. Strains belonging to three other known M. avium serotypes were not included in the phylogeny inference due to apparent lack of the marker sequences in their genomes, as revealed by PCR and Southern blot analysis. These studies suggest that serotypes prevalent in AIDS patients have multiple origins. In trees inferred from both markers, serotype 1 strains, known to have the simplest and shortest GPLs among all other serotypes, were polyphyletic. Likewise, comparisons of the inferred phylogenies with the molecular typing results imply that the existing tools used in epidemiological studies may be poor estimators of M. avium strain relatedness. Additionally, trees inferred from each marker had significantly incongruent topologies due to a well supported alternative placement of strain 2151, suggesting a complex evolutionary history of this genomic region.
-
-
-
Revised description and classification of atypical isolates of Pasteurella multocida from bovine lungs based on genotypic characterization to include variants previously classified as biovar 2 of Pasteurella canis and Pasteurella avium
More LessStrains deviating in key phenotypic characters, mainly isolated from cases of bovine pneumonia in five European countries, were genotyped in order to examine their genotypic relationship with Pasteurella multocida. Twenty-two strains of Pasteurella avium biovar 2, including variants in indole, xylose and mannitol, 18 strains of Pasteurella canis biovar 2 and variants of this taxon, five strains of P. multocida subsp. septica showing variations in indole and ornithine decarboxylase, nine strains of P. multocida subsp. multocida showing variation in ornithine decarboxylase and mannitol, and type strains of the subspecies of P. multocida were included. Ribotyping was used to examine the relationship of the strains, and 13 types, each containing between one and 20 isolates, were observed. Identical ribotypes were observed in some cases for P. avium biovar 2 and either P. canis biovar 2 or P. multocida subsp. septica. ITS (16S–23S rRNA internal transcribed spacer) fragment-length profiling showed identity of the majority of strains (47 of 52), representing all four taxa, with only five divergent strains. A 16S rRNA sequence comparison of 11 strains representing the main ribotype clusters showed 99·9 % similarity to the type strain of P. multocida subsp. multocida, but only 97·4 % similarity was obtained to P. canis (biovar 1) and 93·7 % to P. avium (biovar 1). A species-specific PCR test for P. multocida gave a positive result with biovar 2 variants of P. avium and P. canis. DNA–DNA hybridizations between strains of P. multocida, biovar 2 variants of P. avium and P. canis, and P. multocida subsp. septica confirmed similarity at the species level. It is proposed, on the basis of genotypic similarity, that P. multocida be reclassified to include the biovar 2 variants of P. avium and P. canis and that the existence of the biovar 2 variants of P. avium and P. canis is highly questionable. It is concluded that the redefined P. multocida is genotypically homogeneous, although phenotypically diverse lineages exist with respect to ornithine decarboxylase, indole and mannitol, characters that have been regarded as essential for identification to the species level. A formal reclassification of the species is not possible, however, since too few strains have been found to vary in these key characters. Considering the phenotypic diversity of P. multocida, identification will have to depend partly on genotypic methods and the source host also seems important for safe diagnosis.
-
-
-
Diversity analysis of commensal porcine Escherichia coli – associations between genotypes and habitat in the porcine gastrointestinal tract
More LessDiversity studies of enteric Escherichia coli have relied almost entirely on faecal isolations on the assumption that they are representative of flora found throughout the gastrointestinal tract. The authors have addressed this belief by analysing isolates obtained from the duodenum, ileum, colon and faeces of pigs. E. coli isolates were obtained from eight pigs and characterized using multi-locus enzyme electrophoresis and PCR-based screening for a range of factors thought to be associated with intestinal and extra-intestinal disease. There are four main genetic groups of commensal E. coli (A, B1, B2, D). Group A strains represented 76 % of the isolates from the duodenum, ileum and colon compared to 58 % of the strains isolated from faeces. A nested molecular analysis of variance based on the allozyme and virulence factor screening results showed that differences among individual pigs accounted for 6 % of the observed genetic diversity, whilst 27 % of the genetic variation could be explained by clonal composition differences among gut regions. Finally, the absence of virulence genes in these commensals indicates that they may be suitable as a probiotic consortium, particularly if they also display increased adherence to enterocytes and antagonistic activity against pathogenic strains of E. coli.
-
- CELL AND DEVELOPMENTAL BIOLOGY
-
-
-
Branched swarming patterns on a synthetic medium formed by wild-type Bacillus subtilis strain 3610: detection of different cellular morphologies and constellations of cells as the complex architecture develops
More LessAfter optimizing the conditions, including nutrients and temperature, swarming of Bacillus subtilis 3610 was obtained on a synthetic, fully defined medium. The swarms formed highly branched (dendritic) patterns, generated by successive waves of moving cells. A detailed microscopic in situ analysis of swarms 1 and 2 revealed varied cell morphologies and a remarkable series of events, with cells assembling into different ‘structures’, as the architecture of the swarm developed. Long filamentous cells begin to form before the onset of the first swarming (11 h) and are again observed at later stages in the interior of individual mature dendrites. Swarm 2, detected at 18–22 h, is accompanied by the rapid movement of a wave of dispersed (non-filamentous) cells. Subsequently at the forward edge of this swarm, individual cells begin to cluster together, gradually forming de novo the shape of a dendrite tip with progressive lengthening of this new structure ‘backwards' towards the swarm centre. In both swarms 1 and 2, after the initial clustering of cells, there is the progressive appearance of a spreading monolayer of rafts (4–5 non-filamented cells, neatly aligned). The alternative possible roles of the rafts in the development of the swarm are discussed.
-
-
-
-
Mutant alleles of the essential 14-3-3 gene in Candida albicans distinguish between growth and filamentation
More LessThe opportunistic fungal pathogen Candida albicans has the ability to exploit diverse host environments and can either reside commensally or cause disease. In order to adapt to its new environment it must respond to new physical conditions, nutrient sources, and the host immune response. This requires the co-regulation of multiple signalling networks. The 14-3-3 family of proteins is highly conserved in all eukaryotic species. These proteins regulate signalling pathways involved in cell survival, the cell cycle, and differentiation, and effect their functions via interactions with phosphorylated serines/threonines. In C. albicans there is only one 14-3-3 protein, Bmh1p, and it is required for vegetative growth and optimal filamentation. In order to dissect separate functions of Bmh1p in C. albicans, site-directed nucleotide substitutions were made in the C. albicans BMH1 gene based on studies in other species. Putative temperature-sensitive, ligand-binding and dimerization mutants were constructed. In addition two mutant strains identified through random mutagenesis were analysed. All five mutant strains demonstrated varying defects in growth and filamentation. This paper begins to segregate functions of Bmh1p that are required for optimal growth and the different filamentation pathways. These mutant strains will allow the identification of 14-3-3 target interactions and correlate the individual functions of Bmh1p to cellular processes involved in pathogenesis.
-
- ENVIRONMENTAL MICROBIOLOGY
-
-
-
Diversity and distribution of Microcystis (Cyanobacteria) oligopeptide chemotypes from natural communities studied by single-colony mass spectrometry
More LessMicrocystis sp. has been recognized in recent years as a producer of a high number of secondary metabolites. Among these, peptides that are produced by the non-ribosomal peptide synthetase pathway often show bioactivity or are toxic to humans. The production of particular peptides is specific for individual Microcystis clones, allowing their characterization as chemotypes by analysing the peptidome. The authors studied the in situ diversity of peptides and chemotypes in Microcystis communities from lakes in and around Berlin, Germany, by direct analysis of individual colonies by MALDI-TOF mass spectrometry. From 165 colonies analysed a total of 46 individual peptides could be identified, 21 of which have not been described previously. For six of the new peptides the structures could be elucidated from fragment patterns, while for others only a preliminary classification could be achieved. In most colonies, two to ten individual peptides were detected. In 19 colonies, 16 of which were identified as M. wesenbergii, no peptide metabolites could be detected. The peptide data of 146 colonies were subjected to an ordination (principal components analysis). The principal components were clearly formed by the microcystin variants Mcyst-LR, -RR and -YR, anabaenopeptins B and E/F, a putative microviridin, and a new cyanopeptolin. In the resulting ordination plots most colonies were grouped into five distinct groups, while 40 colonies scattered widely outside these groups. In some cases colonies from different lakes clustered closely, indicating the presence of similar chemotypes in the respective samples. With respect to colony morphology no clear correlation between a chemotype and a morphospecies could be established, but M. aeruginosa, for example, was found to produce predominantly microcystins. In contrast, M. ichthyoblabe colonies were mostly negative for microcystins and instead produced anabaenopeptins. The number of peptides detected in a limited number of samples and the various combinations of peptides in individual Microcystis colonies highlights the immense metabolic potential and diversity of this genus.
-
-
-
-
Characterization of Pseudomonas putida genes responsive to nutrient limitation
More LessThe low bioavailability of nutrients and oxygen in the soil environment has hampered successful expression of biodegradation and biocontrol genes that are driven by promoters highly active during routine laboratory conditions of high availability of nutrients and oxygen. Hence, in the present study, expression of the gus-tagged genes in 12 Tn5-gus mutants of the soil microbe Pseudomonas putida PNL-MK25 were examined under various conditions chosen to mimic the soil environment: low carbon, phosphate, nitrate or oxygen, and in the rhizosphere. Based on their expression profiles, three nutrient-responsive mutant (NRM) strains, NRM5, NRM7 and NRM17, were selected for identification of the tagged genes. In strain NRM5, expression of the glutamate dehydrogenase (gdhA) gene was increased 4·9–26·4-fold under various low-nutrient conditions. In NRM7, expression of the novel NADPH : quinone oxidoreductase-like (nql) gene was consistently amongst the highest and was synergistically upregulated by low-nutrient and anoxic conditions. The cyoD gene in NRM17, which encodes the fourth subunit of the cytochrome o ubiquinol oxidase complex, had decreased expression in low-nutrient conditions but its absolute expression level was still amongst the highest. Additionally, it was independent of oxygen availability, in contrast to that in Escherichia coli.
-
-
-
Multiple linear regression analysis of bacterial deposition to polyurethane coatings after conditioning film formation in the marine environment
More LessMany studies have shown relationships of substratum hydrophobicity, charge or roughness with bacterial adhesion, although bacterial adhesion is governed by interplay of different physico-chemical properties and multiple regression analysis would be more suitable to reveal mechanisms of bacterial adhesion. The formation of a conditioning film of organic compounds adsorbed from seawater affects the properties of substratum surfaces prior to bacterial adhesion, which is a complicating factor in studying the mechanism of bacterial adhesion. In this paper, the impact of conditioning films adsorbed from natural seawater to four polyurethane coatings with different hydrophobicity, elasticity and roughness was studied for three different marine bacterial strains in a multiple linear regression analysis. The water contact angle on hydrophobic coatings decreased on average by 8 degrees and increased on average by the same amount on hydrophilic coatings. These changes were accompanied by increased concentrations of oxygen and nitrogen on the surface as determined by X-ray photoelectron spectroscopy, indicative of adsorption of proteinaceous material. Furthermore, the mean surface roughness increased on average by 4 nm after conditioning film formation. Multiple linear regression analysis revealed that changes in deposition due to conditioning film formation of Marinobacter hydrocarbonoclasticus, Psychrobacter sp. SW5H and Halomonas pacifica in a stagnation-point flow chamber could be explained in a model comprising hydrophobicity and the prevalence of nitrogen-rich components on the surface for the most hydrophobic strain. For the two more hydrophilic strains, deposition was governed by a combination of surface roughness and hydrophobicity. Elasticity was not a factor in bacterial adhesion to conditioning films.
-
-
-
Molecular identification of Vibrio harveyi-related isolates associated with diseased aquatic organisms
More LessFifty strains belonging to Vibrio harveyi, Vibrio campbellii, and the recently described Vibrio rotiferianus, were analysed using phenotypic and genomic techniques with the aim of analysing the usefulness of the different techniques for the identification of V. harveyi-related species. The species V. harveyi and V. campbellii were phenotypically indistinguishable by more than 100 phenotypic features. Thirty-nine experimental strains were phenotypically identified as V. harveyi, but FAFLP, REP-PCR, IGS-PCR and DNA–DNA hybridization proved that they in fact belong to the species V. campbellii. Similar groupings were found among all fingerprinting methodologies (except IGS-PCR). Thirty-two experimental strains clustered with the V. campbellii type and one reference strain; seven strains clustered with the V. harveyi type and three reference strains; and the type and four reference strains of V. rotiferianus grouped together. The correlations between DNA–DNA hybridization and the genomic fingerprinting by FAFLP and (GTG)5-PCR were found to be above 0·68 and statistically significant, suggesting the value of the latter techniques for the reliable identification of V. harveyi-related species. The results presented indicate that strains phenotypically identified as V. harveyi are in fact V. campbellii; these findings position V. campbellii as an important species involved in diseases of reared aquatic organisms.
-
- GENES AND GENOMES
-
-
-
Naturally occurring horizontal gene transfer and homologous recombination in Mycobacterium
More LessAcquisition of genetic information through horizontal gene transfer (HGT) is an important evolutionary process by which micro-organisms gain novel phenotypic characteristics. In pathogenic bacteria, for example, it facilitates maintenance and enhancement of virulence and spread of drug resistance. In the genus Mycobacterium, to which several primary human pathogens belong, HGT has not been clearly demonstrated. The few existing reports suggesting this process are based on circumstantial evidence of similarity of sequences found in distantly related species. Here, direct evidence of HGT between strains of Mycobacterium avium representing two different serotypes is presented. Conflicting evolutionary histories of genes encoding elements of the glycopeptidolipid (GPL) biosynthesis pathway led to an analysis of the GPL cluster genomic sequences from four Mycobacterium avium strains. The sequence of M. avium strain 2151 appeared to be a mosaic consisting of three regions having alternating identities to either M. avium strains 724 or 104. Maximum-likelihood estimation of two breakpoints allowed a ∼4100 bp region horizontally transferred into the strain 2151 genome to be pinpointed with confidence. The maintenance of sequence continuity at both breakpoints and the lack of insertional elements at these sites strongly suggest that the integration of foreign DNA occurred by homologous recombination. To our knowledge, this is the first report to demonstrate naturally occurring homologous recombination in Mycobacterium. This previously undiscovered mechanism of genetic exchange may have major implications for the understanding of Mycobacterium pathogenesis.
-
-
-
-
GvpE- and GvpD-mediated transcription regulation of the p-gvp genes encoding gas vesicles in Halobacterium salinarum
More LessThe transcription of the 14 p-gvp genes involved in gas vesicle formation of Halobacterium salinarum PHH1 is driven by the four promoters pA, pD, pF and pO. The regulation of these promoters was investigated in Haloferax volcanii transformants with respect to the endogenous regulatory proteins GvpE and GvpD. Northern analyses demonstrated that the transcription derived from the pA and pD promoters was enhanced by GvpE, whereas the activities of the pF and pO promoters were not affected. Similar results were obtained using promoter fusions with the bgaH reporter gene encoding an enzyme with β-galactosidase activity. The largest amount of specific β-galactosidase activity was determined for pA-bgaH transformants, followed by pF-bgaH and pD-bgaH transformants. The presence of GvpE resulted in a severalfold induction of the pA and pD promoter, whereas the pF promoter was not affected. A lower GvpE-induced pA promoter activity was seen in the presence of GvpD in the pA-bgaH/DEex transformants, suggesting a function of GvpD in repression. To determine the DNA sequences involved in the GvpE-mediated activation, a 50-nucleotide region of the pA promoter was investigated by 4-nucleotide scanning mutagenesis. Some of these mutations affected the basal transcription, especially mutations in the region of the TATA box and the putative BRE sequence element, and also around position −10. Mutant E, harbouring a sequence with greater identity to the consensus BRE element, showed a significantly enhanced basal promoter activity compared to wild-type. Mutations not affecting basal transcription, but yielding a reduced GvpE-mediated activation, were located immediately upstream of BRE. These results suggested that the transcription activation by GvpE is in close contact with the core transcription machinery.
-
-
-
Sequence analysis of two plasmids from the phytoplasma beet leafhopper-transmitted virescence agent
More LessThe complete nucleotide sequences of the two plasmids from the phytoplasma beet leafhopper-transmitted virescence agent (BLTVA) have been determined. The larger plasmid, pBLTVA-1, was 10 785 nt in length and contained 11 putative ORFs, almost all of which were duplicated or triplicated on the plasmid due to the presence of large repeated regions. The sequence contained a series of tandem repeats, the largest of which was 338 nt long. The sequences of ORFs 4 and 11 showed homology with the replication genes of plasmids from other phytoplasmas and from geminiviruses. ORF9, the only ORF present as a single copy, showed homology with DNA primase genes from bacterial chromosomes and contained the conserved zinc finger and topoisomerase/primase domains. None of the other eight ORFs showed homology with known sequences in the GenBank database. pBLTVA-2 was 2587 nt in length, and all of its sequence was nearly identical to sequences from pBLTVA-1, most of which spanned ORFs 10 and 11, including the 338 nt tandem repeat. Analysis of 30 strains of BLTVA showed that most of the 11 putative ORFs were present, but the size of the plasmids varied in these strains.
-
- MICROBIOLOGY COMMENT
-
- PATHOGENS AND PATHOGENICITY
-
-
-
The Campylobacter jejuni general glycosylation system is important for attachment to human epithelial cells and in the colonization of chicks
More LessIt has recently been shown that the enteropathogen Campylobacter jejuni has an N-linked general protein glycosylation pathway (Pgl) that modifies many of the organism's proteins. To determine the role of the N-linked general glycosylation in C jejuni, the authors studied the pglH gene, which shows high similarity to a family of sugar transferases. pglH mutants were constructed in strains 81116 and 11168H. Both mutants were shown to be deficient in their ability to glycosylate a number of C. jejuni proteins, but their lipooligosaccharide and capsule were unaffected. The pglH mutants had significantly reduced ability to adhere to and invade human epithelial Caco-2 cells. Additionally, the 81116 pglH mutant was severely affected in its ability to colonize chicks. These results suggest that glycosylation is important for the attachment of C. jejuni to human and chicken host cells and imply a role for glycoproteins in the pathogenesis of C. jejuni.
-
-
-
-
Molecular cloning of haemoglobin-binding protein HgbA in the outer membrane of Actinobacillus pleuropneumoniae
More LessFrom the porcine pathogen Actinobacillus pleuropneumoniae cultivated in iron-deficient or haem-deficient media, haemoglobin (Hb)-agarose affinity purification was exploited to isolate an outer-membrane protein of ∼105 kDa, designated HgbA. Internal peptide sequences of purified HgbA were used to design oligonucleotide primers for PCR amplification, yielding amplicons that showed partial sequences with homology to hgbA of Pasteurella multocida. Upon screening two genomic libraries of A. pleuropneumoniae serotype 1 strain 4074, positive clones were assembled into an ORF of 2838 bp. HgbA (946 aa) includes a signal peptide of 23 aa and the deduced HgbA sequence (104 890 Da) also demonstrated a possible Ton box. The promoter region of hgbA from A. pleuropneumoniae serotype 1 showed consensus for −35 and −10 sequences and a putative Fur-binding site. RT-PCR confirmed that hgbA of A. pleuropneumoniae is upregulated in response to diminished levels of iron in the culture medium. While an internally deleted hgbA mutant was unable to use pig Hb as sole source of iron for growth, flow cytometry confirmed its Hb binding; the internally deleted sequences may not be required for Hb binding, but appear necessary for the iron supply from Hb. HgbA is required for growth of A. pleuropneumoniae in the presence of Hb as sole iron source.
-
-
-
Dosage-dependent functions of fatty acid desaturase Ole1p in growth and morphogenesis of Candida albicans
More LessConditions in the infected human host trigger virulence attributes of the fungal pathogen Candida albicans. Specific inducers and elevated temperatures lead to hyphal development or regulate chlamydospore development. To explore if these processes are affected by membrane lipids, an investigation of the functions of the Ole1 fatty acid desaturase (stearoyl-CoA desaturase) in C. albicans, which synthesizes oleic acid, was undertaken. A conditional strain expressing OLE1 from the regulatable MET3 promoter was unable to grow in repressing conditions, indicating that OLE1 is an essential gene. In contrast, a mutant lacking both alleles of OLE2, encoding a Ole1p homologue, was viable and had no apparent phenotypes. Partial repression of MET3p–OLE1 slightly lowered oleic acid levels and decreased membrane fluidity; these conditions permitted growth in the yeast form, but prevented hyphal development in aerobic conditions and blocked the formation of chlamydospores. In contrast, in hypoxic conditions, which trigger an alternative morphogenetic pathway, hyphal morphogenesis was unaffected. Because aerobic morphogenetic signalling and oleic acid biosynthesis require oxygen, it is proposed that oleic acid may function as a sensor activating specific morphogenetic pathways in normoxic conditions.
-
-
-
The hyaluronate lyase of Staphylococcus aureus – a virulence factor?
More LessThe hyaluronate lyase (HL) gene of Staphylococcus aureus 8325-4 (hysA) was inactivated in vitro with the insertion of the erythromycin determinant, ermC, from plasmid pE194. The hysA : : ermC mutation was introduced into S. aureus via a temperature-sensitive shuttle vector, where it underwent homologous recombination with the wild-type (w.t.) allele. The insertion of ermC in the chromosomal hysA locus was confirmed by Southern blot hybridization and the loss of HL activity was demonstrated macroscopically by a plate assay. The importance of HL for pathogenicity was assessed by comparing the virulence of the HL− mutant strain to that of the w.t. in an established mouse abscess model of S. aureus infection. A significantly higher cell recovery was obtained from lesions infected with the w.t. strain compared to the lesions infected with the HL− strain (P =0·01). Although the lesion areas from both groups were not significantly different (P=0·9) they were of different morphology. A colorimetric assay was used to measure HL activity from culture supernatants of the S. aureus 8325-4 strains w.t., WA250 (agr) and PC1839 (sar) grown in a chemically defined medium. HL activity reached a maximum in the w.t. strain during mid-exponential phase (t=5 h) and while it showed a 16-fold decrease in the agr mutant it increased 35-fold in the sar mutant background. These results strongly suggest that HL is a virulence factor which is important in the early stages of subcutaneous infections.
-
-
-
Oxidative and amphotericin B-mediated cell death in the opportunistic pathogen Aspergillus fumigatus is associated with an apoptotic-like phenotype
More LessWhen protoplasts of the opportunistic fungal pathogen Aspergillus fumigatus were treated with low but toxic levels of hydrogen peroxide (0·1 mM) or amphotericin B (0·5 μg ml−1), loss of cell viability and death were associated with a number of phenotypic changes characteristic of apoptosis. The percentage of protoplasts staining positive with annexin V-FITC, an indicator of the externalization of phosphatidylserine and an early marker of apoptosis, rose to ∼55 % within 1 h. This was followed by a similar increase in apoptotic DNA fragmentation detected by the TUNEL assay, and led to a loss of cell permeability and death in ∼90 % of protoplasts, as indicated by the uptake of propidium iodide. The development of an apoptotic phenotype was blocked when protoplasts were pre-treated with the protein synthesis inhibitor cycloheximide, indicating active participation of the cell in the process. However, no significant activity against synthetic caspase substrates was detected, and the inclusion of the cell-permeant broad-spectrum caspase inhibitor Z-VAD-fmk did not block the development of the apoptotic-like phenotype. Higher concentrations of H2O2 (1·8 mM) and amphotericin B (1 μg ml−1) caused protoplasts to die without inducing an apoptotic phenotype. As predicted, the fungistatic antifungal agent itraconazole, which inhibits growth without causing immediate cell death, did not induce an apoptotic-like phenotype.
-
- PHYSIOLOGY
-
-
-
The transcription of the cbb operon in Nitrosomonas europaea
More LessNitrosomonas europaea is an aerobic ammonia-oxidizing bacterium that participates in the C and N cycles. N. europaea utilizes CO2 as its predominant carbon source, and is an obligate chemolithotroph, deriving all the reductant required for energy and biosynthesis from the oxidation of ammonia (NH3) to nitrite (
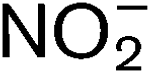 ). This bacterium fixes carbon via the Calvin–Benson–Bassham (CBB) cycle via a type I ribulose bisphosphate carboxylase/oxygenase (RubisCO). The RubisCO operon is composed of five genes, cbbLSQON. This gene organization is similar to that of the operon for ‘green-like’ type I RubisCOs in other organisms. The cbbR gene encoding the putative regulatory protein for RubisCO transcription was identified upstream of cbbL. This study showed that transcription of cbb genes was upregulated when the carbon source was limited, while amo, hao and other energy-harvesting-related genes were downregulated. N. europaea responds to carbon limitation by prioritizing resources towards key components for carbon assimilation. Unlike the situation for amo genes, NH3 was not required for the transcription of the cbb genes. All five cbb genes were only transcribed when an external energy source was provided. In actively growing cells, mRNAs from the five genes in the RubisCO operon were present at different levels, probably due to premature termination of transcription, rapid mRNA processing and mRNA degradation.
). This bacterium fixes carbon via the Calvin–Benson–Bassham (CBB) cycle via a type I ribulose bisphosphate carboxylase/oxygenase (RubisCO). The RubisCO operon is composed of five genes, cbbLSQON. This gene organization is similar to that of the operon for ‘green-like’ type I RubisCOs in other organisms. The cbbR gene encoding the putative regulatory protein for RubisCO transcription was identified upstream of cbbL. This study showed that transcription of cbb genes was upregulated when the carbon source was limited, while amo, hao and other energy-harvesting-related genes were downregulated. N. europaea responds to carbon limitation by prioritizing resources towards key components for carbon assimilation. Unlike the situation for amo genes, NH3 was not required for the transcription of the cbb genes. All five cbb genes were only transcribed when an external energy source was provided. In actively growing cells, mRNAs from the five genes in the RubisCO operon were present at different levels, probably due to premature termination of transcription, rapid mRNA processing and mRNA degradation.
-
-
-
-
Specific growth rate and not cell density controls the general stress response in Escherichia coli
More LessIn batch cultures of Escherichia coli, the intracellular concentration of the general stress response sigma factor RpoS typically increases during the transition from the exponential to the stationary growth phase. However, because this transition is accompanied by complex physico-chemical and biological changes, which signals predominantly elicit this induction is still the subject of debate. Careful design of the growth environment in chemostat and batch cultures allowed the separate study of individual factors affecting RpoS. Specific growth rate, and not cell density or the nature of the growth-limiting nutrient, controlled RpoS expression and RpoS-dependent hydroperoxidase activity. Furthermore, it was demonstrated that the standard E. coli minimal medium A (MMA) is not suitable for high-cell-density cultivation because it lacks trace elements. Previously reported cell-density effects in chemostat cultures of E. coli can be explained by a hidden, secondary nutrient limitation, which points to the importance of medium design and appropriate experimental set-up for studying cell-density effects.
-
-
-
Formation of ‘non-culturable’ cells of Mycobacterium smegmatis in stationary phase in response to growth under suboptimal conditions and their Rpf-mediated resuscitation
More LessConditions were investigated that promote the formation of ‘non-culturable’ (NC) cells of Mycobacterium (Myc.) smegmatis in stationary phase. After cultivation in a rich medium, or under conditions that may be considered optimal for bacterial growth, or starvation for carbon, nitrogen or phosphorus, bacteria failed to enter a NC state. However, when grown under suboptimal conditions, resulting in a reduced growth rate or maximal cell concentration (e.g. in modified Hartman's–de Bont medium), bacteria adopted a stable NC state after 3–4 days incubation in stationary phase. Such conditions are not specific as purF and devR mutants of Myc. smegmatis also showed (transient) loss of culturability following growth to stationary phase in an optimized medium, but under oxygen-limited conditions. The behaviour of the same mutants in oxygen-sufficient but nutrient-inappropriate medium (modified Hartman's–de Bont medium) was similar to that of the wild-type (adoption of a stable NC state). It is hypothesized that adoption of a NC state may represent an adaptive response of the bacteria, grown under conditions when their metabolism is significantly compromised due to the simultaneous action of several factors, such as usage of inappropriate nutrients or low oxygen availability or impairment of a particular metabolic pathway. NC cells of wild-type Myc. smegmatis resume growth when transferred to a suitable resuscitation medium. Significantly, resuscitation was observed when either recombinant Rpf protein or supernatant derived from a growing bacterial culture was incorporated into the resuscitation medium. Moreover, co-culture with Micrococcus (Mcc.) luteus cells (producing and secreting Rpf) also permitted resuscitation. Isogenic strains of Myc. smegmatis harbouring plasmids containing the Mcc. luteus rpf gene also adopt a similar NC state after growth to stationary phase in modified Hartman's–de Bont medium. However, in contrast to the behaviour noted above, these strains resuscitated spontaneously when transferred to the resuscitation medium, presumably because they are able to resume endogenous synthesis of Mcc. luteus Rpf. Resuscitation was not observed in the control strain harbouring a plasmid lacking Mcc. luteus rpf. In contrast to wild-type, the NC cells of purF and devR mutants obtained under oxygen-limited conditions resuscitate spontaneously, presumably because the heterogeneous population contains some residual viable cells that continue to make Rpf-like proteins.
-
-
-
Phototrophic utilization of taurine by the purple nonsulfur bacteria Rhodopseudomonas palustris and Rhodobacter sphaeroides
More LessTaurine metabolism by two phototrophically grown purple nonsulfur bacteria enrichment isolates has been examined. Rhodopseudomonas palustris (strain Tau1) grows with taurine as a sole electron donor, sulfur and nitrogen source during photoautotrophic growth. Rhodobacter sphaeroides (strain Tau3) grows on the compound as sole electron donor, sulfur and nitrogen source, and partial carbon source, in the presence of CO2 during photoheterotrophic growth. Both organisms utilize an inducible taurine–pyruvate aminotransferase and a sulfoacetaldehyde acetyltransferase. The products of this metabolism are bisulfite and acetyl phosphate. Bisulfite ultimately was oxidized to sulfate, but this was not an adequate source of electrons for photometabolism. Experiments using either [U-14C]taurine or 14CO2 demonstrated that Rb. sphaeroides Tau3 assimilated the carbon from approximately equimolar amounts of taurine and exogenous CO2. The taurine-carbon assimilation was not diminished by excess non-radioactive bicarbonate. Malate synthase (but not isocitrate lyase) was induced in these taurine-grown cells. It is concluded that assimilation of taurine carbon occurs through an intermediate other than CO2. Similar labelling experiments with Rp. palustris Tau1 determined that taurine is utilized only as an electron donor for the reduction of CO2, which contributes all the cell carbon. Photoautotrophic metabolism was confirmed in this organism by the absence of either malate synthase or isocitrate lyase in taurine+CO2-grown cells. Culture collection strains of these two bacteria did not utilize taurine in these fashions.
-
-
-
Oxidative stress response in Clostridium perfringens
More LessClostridium perfringens, a strictly anaerobic bacterium, is able to survive when exposed to oxygen for short periods of time and exhibits a complex adaptive response to reactive oxygen species, both under aerobic and anaerobic conditions. However, this adaptive response is not completely understood. C. perfringens possesses specialized genes that might be involved in this adaptive process, such as those encoding superoxide dismutase (SOD), superoxide reductase and alkyl hydroperoxide reductase, but their contribution to the oxidative stress response and their control mechanisms are unknown. By a combination of functional complementation of Escherichia coli strains impaired in either SOD, alkyl hydroperoxide reductase (AhpC) or catalase activity (Cat), transcription analysis and characterization of mutants impaired in regulatory genes, it was concluded that: (i) the product of the sod gene is certainly essential to scavenge superoxide radicals, (ii) the ahpC gene, which is fully induced in all oxidative stress conditions, is probably involved in the scavenging of all intracellular peroxides, (iii) the three rubrerythrin (rbr) genes of C. perfringens do not encode proteins with in vivo H2O2 reductase activity, and (iv) the two rubredoxin (rub) genes do not contribute to the hypothetical superoxide reductase activity, but are likely to belong to an electron transfer chain involved in energy metabolism.
-
- REVIEWS
-
-
-
The replication-related organization of bacterial genomes
More LessThe replication of the chromosome is among the most essential functions of the bacterial cell and influences many other cellular mechanisms, from gene expression to cell division. Yet the way it impacts on the bacterial chromosome was not fully acknowledged until the availability of complete genomes allowed one to look upon genomes as more than bags of genes. Chromosomal replication includes a set of asymmetric mechanisms, among which are a division in a lagging and a leading strand and a gradient between early and late replicating regions. These differences are the causes of many of the organizational features observed in bacterial genomes, in terms of both gene distribution and sequence composition along the chromosome. When asymmetries or gradients increase in some genomes, e.g. due to a different composition of the DNA polymerase or to a higher growth rate, so do the corresponding biases. As some of the features of the chromosome structure seem to be under strong selection, understanding such biases is important for the understanding of chromosome organization and adaptation. Inversely, understanding chromosome organization may shed further light on questions relating to replication and cell division. Ultimately, the understanding of the interplay between these different elements will allow a better understanding of bacterial genetics and evolution.
-
-
-
-
Substrate recognition by nonribosomal peptide synthetase multi-enzymes
More LessNonribosomal peptide synthetases (NRPSs) are giant multi-domain enzymes that catalyse the biosynthesis of many commercially important peptides produced by bacteria and fungi. Several studies over the last decade have shown that many of the individual domains within NRPSs exhibit significant substrate selectivity, which impacts on our ability to engineer NRPSs to produce new bioactive microbial peptides. Adenylation domains appear to be the primary determinants of substrate selectivity in NRPSs. Much progress has been made towards an empirical understanding of substrate selection by these domains over the last 5 years, but the molecular basis of substrate selectivity in these domains is not yet well understood. Perhaps surprisingly, condensation domains have also been reported to exhibit moderate to high substrate selectivity, although the generality of this observation and its potential impact on engineered biosynthesis experiments has yet to be fully elucidated. The situation is less clear for the thioesterase domains, which seem in certain cases to be dedicated to the hydrolysis/cyclization of their natural substrate, whereas in other cases they are largely permissive.
-
Volumes and issues
-
Volume 170 (2024)
-
Volume 169 (2023)
-
Volume 168 (2022)
-
Volume 167 (2021)
-
Volume 166 (2020)
-
Volume 165 (2019)
-
Volume 164 (2018)
-
Volume 163 (2017)
-
Volume 162 (2016)
-
Volume 161 (2015)
-
Volume 160 (2014)
-
Volume 159 (2013)
-
Volume 158 (2012)
-
Volume 157 (2011)
-
Volume 156 (2010)
-
Volume 155 (2009)
-
Volume 154 (2008)
-
Volume 153 (2007)
-
Volume 152 (2006)
-
Volume 151 (2005)
-
Volume 150 (2004)
-
Volume 149 (2003)
-
Volume 148 (2002)
-
Volume 147 (2001)
-
Volume 146 (2000)
-
Volume 145 (1999)
-
Volume 144 (1998)
-
Volume 143 (1997)
-
Volume 142 (1996)
-
Volume 141 (1995)
-
Volume 140 (1994)
-
Volume 139 (1993)
-
Volume 138 (1992)
-
Volume 137 (1991)
-
Volume 136 (1990)
-
Volume 135 (1989)
-
Volume 134 (1988)
-
Volume 133 (1987)
-
Volume 132 (1986)
-
Volume 131 (1985)
-
Volume 130 (1984)
-
Volume 129 (1983)
-
Volume 128 (1982)
-
Volume 127 (1981)
-
Volume 126 (1981)
-
Volume 125 (1981)
-
Volume 124 (1981)
-
Volume 123 (1981)
-
Volume 122 (1981)
-
Volume 121 (1980)
-
Volume 120 (1980)
-
Volume 119 (1980)
-
Volume 118 (1980)
-
Volume 117 (1980)
-
Volume 116 (1980)
-
Volume 115 (1979)
-
Volume 114 (1979)
-
Volume 113 (1979)
-
Volume 112 (1979)
-
Volume 111 (1979)
-
Volume 110 (1979)
-
Volume 109 (1978)
-
Volume 108 (1978)
-
Volume 107 (1978)
-
Volume 106 (1978)
-
Volume 105 (1978)
-
Volume 104 (1978)
-
Volume 103 (1977)
-
Volume 102 (1977)
-
Volume 101 (1977)
-
Volume 100 (1977)
-
Volume 99 (1977)
-
Volume 98 (1977)
-
Volume 97 (1976)
-
Volume 96 (1976)
-
Volume 95 (1976)
-
Volume 94 (1976)
-
Volume 93 (1976)
-
Volume 92 (1976)
-
Volume 91 (1975)
-
Volume 90 (1975)
-
Volume 89 (1975)
-
Volume 88 (1975)
-
Volume 87 (1975)
-
Volume 86 (1975)
-
Volume 85 (1974)
-
Volume 84 (1974)
-
Volume 83 (1974)
-
Volume 82 (1974)
-
Volume 81 (1974)
-
Volume 80 (1974)
-
Volume 79 (1973)
-
Volume 78 (1973)
-
Volume 77 (1973)
-
Volume 76 (1973)
-
Volume 75 (1973)
-
Volume 74 (1973)
-
Volume 73 (1972)
-
Volume 72 (1972)
-
Volume 71 (1972)
-
Volume 70 (1972)
-
Volume 69 (1971)
-
Volume 68 (1971)
-
Volume 67 (1971)
-
Volume 66 (1971)
-
Volume 65 (1971)
-
Volume 64 (1970)
-
Volume 63 (1970)
-
Volume 62 (1970)
-
Volume 61 (1970)
-
Volume 60 (1970)
-
Volume 59 (1969)
-
Volume 58 (1969)
-
Volume 57 (1969)
-
Volume 56 (1969)
-
Volume 55 (1969)
-
Volume 54 (1968)
-
Volume 53 (1968)
-
Volume 52 (1968)
-
Volume 51 (1968)
-
Volume 50 (1968)
-
Volume 49 (1967)
-
Volume 48 (1967)
-
Volume 47 (1967)
-
Volume 46 (1967)
-
Volume 45 (1966)
-
Volume 44 (1966)
-
Volume 43 (1966)
-
Volume 42 (1966)
-
Volume 41 (1965)
-
Volume 40 (1965)
-
Volume 39 (1965)
-
Volume 38 (1965)
-
Volume 37 (1964)
-
Volume 36 (1964)
-
Volume 35 (1964)
-
Volume 34 (1964)
-
Volume 33 (1963)
-
Volume 32 (1963)
-
Volume 31 (1963)
-
Volume 30 (1963)
-
Volume 29 (1962)
-
Volume 28 (1962)
-
Volume 27 (1962)
-
Volume 26 (1961)
-
Volume 25 (1961)
-
Volume 24 (1961)
-
Volume 23 (1960)
-
Volume 22 (1960)
-
Volume 21 (1959)
-
Volume 20 (1959)
-
Volume 19 (1958)
-
Volume 18 (1958)
-
Volume 17 (1957)
-
Volume 16 (1957)
-
Volume 15 (1956)
-
Volume 14 (1956)
-
Volume 13 (1955)
-
Volume 12 (1955)
-
Volume 11 (1954)
-
Volume 10 (1954)
-
Volume 9 (1953)
-
Volume 8 (1953)
-
Volume 7 (1952)
-
Volume 6 (1952)
-
Volume 5 (1951)
-
Volume 4 (1950)
-
Volume 3 (1949)
-
Volume 2 (1948)
-
Volume 1 (1947)
Most Read This Month
