 -
Volume 152,
Issue 7,
2006
-
Volume 152,
Issue 7,
2006

Volume 152, Issue 7, 2006
- Microbiology Comment
-
- Cell And Developmental Biology
-
-
-
Ultrastructure and gliding motility of Mycoplasma amphoriforme, a possible human respiratory pathogen
More LessDespite their small size and reduced genomes, many mycoplasma cells have complex structures involved in virulence. Mycoplasma pneumoniae has served as a model for the study of virulence factors of a variety of mycoplasma species that cause disease in humans and animals. These cells feature an attachment organelle, which mediates cytadherence and gliding motility and is required for virulence. An essential component of the architecture of the attachment organelle is an internal detergent-insoluble structure, the electron-dense core. Little information is known regarding its underlying mechanisms. Mycoplasma amphoriforme, a close relative of both M. pneumoniae and the avian pathogen Mycoplasma gallisepticum, is a recently discovered organism associated with chronic bronchitis in immunosuppressed individuals. This work describes both the ultrastructure of M. amphoriforme strain A39T as visualized by scanning electron microscopy and the gliding motility characteristics of this organism on glass. Though externally resembling M. gallisepticum, M. amphoriforme cells were found to have a Triton X-100-insoluble structure similar to the M. pneumoniae electron-dense core but with different dimensions. M. amphoriforme also exhibited gliding motility using time-lapse microcinematography; its movement was slower than that of either M. pneumoniae or M. gallisepticum.
-
-
- Biochemistry And Molecular Biology
-
-
-
Metabolic and regulatory engineering of Serratia marcescens: mimicking phage-mediated horizontal acquisition of antibiotic biosynthesis and quorum-sensing capacities
More LessSerratia marcescens is an important cause of opportunistic human infections. Many, but not all, strains produce prodigiosin, a secondary metabolic, red-pigment antibiotic, the biosynthesis of which is directed by the pig gene cluster. Quorum sensing (QS) involves the production and detection of chemical signal molecules as a means to regulate gene expression in response to population cell density. Several strains of S. marcescens have previously been shown to possess an N-acyl-l-homoserine lactone (aHSL) QS system. This study aimed to determine the impact of introducing, by phage-mediated horizontal gene transfer, a biosynthetic gene cluster (pig) and a regulatory locus (aHSL QS) into strains lacking the respective trait. The pig cluster from S. marcescens ATCC 274 (Sma 274) was transferred to the non-pigmented strain, S. marcescens strain 12 (Sma 12). In the engineered strain, pigment was expressed and brought under the control of the recipient's native regulatory systems (aHSL QS and luxS). Moreover, transfer of the aHSL locus from Sma 12 to the non-QS Sma 274 resulted in the imposition of aHSL control onto a variety of native traits, including pigment production. In addition, during this study, the QS regulon of the clinical strain, Sma 12, was characterized, and some novel QS-regulated traits in S. marcescens were identified. The results have implications for the evolution and dissemination of biosynthetic and QS loci, illustrating the genetic modularity and ease of acquisition of these traits and the capacity of phages to act as vectors for horizontal gene transfer.
-
-
-
-
Broad-spectrum antibacterial activity by a novel abiogenic peptide mimic
More LessThe human-mediated use and abuse of classical antibiotics has created a strong selective pressure for the rapid evolution of antibiotic resistance. As resistance levels rise, and the efficacy of classical antibiotics wanes, the intensity of the search for alternative antimicrobials has increased. One class of molecules that has attracted much attention is the antimicrobial peptides (AMPs). They exhibit broad-spectrum activity, they are potent and they are widespread as part of the innate defence system of both vertebrates and invertebrates. However, peptides are complex molecules that suffer from proteolytic degradation. The ability to capture the essential properties of antimicrobial peptides in simple easy-to-prepare molecules that are abiotic in origin and non-proteolytic offers many advantages. Mechanistic and structural knowledge of existing AMPs was used to design a novel compound that mimics the biochemical activity of an AMP. This report describes the development and in vitro characterization of a small peptide mimic that exhibited quick-acting and selective antibacterial activity against a broad range of bacteria, including numerous clinically relevant strains, at low MIC values.
-
-
-
Identification and functional analysis of the genes for naphthalenesulfonate catabolism by Sphingomonas xenophaga BN6
More LessSphingomonas xenophaga BN6 degrades various (substituted) naphthalenesulfonates to the corresponding (substituted) salicylates. A gene cluster was identified on the plasmid pBN6 which coded for several enzymes participating in the degradative pathway for naphthalenesulfonates. A DNA fragment of 16 915 bp was sequenced which contained 17 ORFs. The genes encoding the 1,2-dihydroxynaphthalene dioxygenase, 2-hydroxychromene-2-carboxylate isomerase, and 2′-hydroxybenzalpyruvate aldolase of the naphthalenesulfonate pathway were identified on the DNA fragment and the encoded proteins heterologously expressed in Escherichia coli. Also, the genes encoding the ferredoxin and ferredoxin reductase of a multi-component, ring-hydroxylating naphthalenesulfonate dioxygenase were identified by insertional inactivation. The identified genes generally demonstrated the highest degree of homology to enzymes encoded by the phenanthrene-degrading organism Sphingomonas sp. P2, or the megaplasmid pNL1 of the naphthalene- and biphenyl-degrading strain Sphingomonas aromaticivorans F199. The genes of S. xenophaga BN6 participating in the degradation of naphthalenesulfonates also shared the same organization in three different transcriptional units as the genes involved in the degradation of naphthalene, biphenyl, and phenanthrene previously found in Sphingomonas sp. P2 and S. aromaticivorans F199. The genes were flanked in S. xenophaga BN6 by ORFs which specify proteins that show the highest homologies to proteins of mobile genetic elements.
-
-
-
Molecular characterization of a conserved archaeal copper resistance (cop) gene cluster and its copper-responsive regulator in Sulfolobus solfataricus P2
More LessUsing a comparative genomics approach, a copper resistance gene cluster has been identified in multiple archaeal genomes. The cop cluster is predicted to encode a metallochaperone (CopM), a P-type copper-exporting ATPase (CopA) and a novel, archaea-specific transcriptional regulator (CopT) which might control the expression of the cop genes. Sequence analysis revealed that CopT has an N-terminal DNA-binding helix–turn–helix domain and a C-terminal TRASH domain; TRASH is a novel domain which has recently been proposed to be uniquely involved in metal-binding in sensors, transporters and trafficking proteins in prokaryotes. The present study describes the molecular characterization of the cop gene cluster in the thermoacidophilic crenarchaeon Sulfolobus solfataricus. The polycistronic copMA transcript was found to accumulate in response to growth-inhibiting copper concentrations, whereas copT transcript abundance appeared to be constitutive. DNA-binding assays revealed that CopT binds to the copMA promoter at multiple sites, both upstream and downstream of the predicted TATA-BRE site. Copper was found to specifically modulate the affinity of DNA binding by CopT. This study describes a copper-responsive operon in archaea, a new family of archaeal DNA-binding proteins, and supports the idea that this domain plays a prominent role in the archaeal copper response. A model is proposed for copper-responsive transcriptional regulation of the copMA gene cluster.
-
-
-
Expression of the Giardia lamblia cyst wall protein 2 in Lactococcus lactis
More LessIn this study, Lactococcus lactis was engineered to express Giardia lamblia cyst wall protein 2 (CWP2) at three different subcellular locations, intracellular, secreted or cell-surface-anchored, using nisin as an inducing agent. CWP2 expression did not appear to be detrimental to L. lactis viability. No particular subcellular location of CWP2 expression offered any advantages over the others with respect to decreased toxicity towards the bacteria. All recombinant lactococci experienced a similar reduction in growth rate when induced. It was determined whether recombinant lactococcal cells engineered for cell surface expression of CWP2 were capable of inducing a CWP2-specific mucosal IgA antibody response. Recombinant lactococci were successful at inducing CWP2-specific IgA antibodies. Moreover, in a pilot challenge experiment, mice immunized with these recombinant lactococci demonstrated a significant (63 %) reduction in cyst output. Thus, it has been demonstrated that G. lamblia CWP2 may be expressed in L. lactis and that recombinant lactococcal cells elicit Giardia-specific antibodies which reduce cyst shedding in a murine model.
-
-
-
Dysgalacticin: a novel, plasmid-encoded antimicrobial protein (bacteriocin) produced by Streptococcus dysgalactiae subsp. equisimilis
More LessDysgalacticin is a novel bacteriocin produced by Streptococcus dysgalactiae subsp. equisimilis strain W2580 that has a narrow spectrum of antimicrobial activity directed primarily against the principal human streptococcal pathogen Streptococcus pyogenes. Unlike many previously described bacteriocins of Gram-positive bacteria, dysgalacticin is a heat-labile 21.5 kDa anionic protein that kills its target without inducing lysis. The N-terminal amino acid sequence of dysgalacticin [Asn-Glu-Thr-Asn-Asn-Phe-Ala-Glu-Thr-Gln-Lys-Glu-Ile-Thr-Thr-Asn-(Asn)-Glu-Ala] has no known homologue in publicly available sequence databases. The dysgalacticin structural gene, dysA, is located on the indigenous plasmid pW2580 of strain W2580 and encodes a 220 aa preprotein which is probably exported via a Sec-dependent transport system. Natural dysA variants containing conservative amino acid substitutions were also detected by sequence analyses of dysA elements from S. dysgalactiae strains displaying W2580-like inhibitory profiles. Production of recombinant dysgalacticin by Escherichia coli confirmed that this protein is solely responsible for the inhibitory activity exhibited by strain W2580. A combination of in silico secondary structure prediction and reductive alkylation was employed to demonstrate that dysgalacticin has a novel structure containing a disulphide bond essential for its biological activity. Moreover, dysgalacticin displays similarity in predicted secondary structure (but not primary amino acid sequence or inhibitory spectrum) with another plasmid-encoded streptococcal bacteriocin, streptococcin A-M57 from S. pyogenes, indicating that dysgalacticin represents a prototype of a new class of antimicrobial proteins.
-
-
-
A screening system for carbon sources enhancing β-N-acetylglucosaminidase formation in Hypocrea atroviridis (Trichoderma atroviride)
More LessTo identify carbon sources that trigger β-N-acetylglucosaminidase (NAGase) formation in Hypocrea atroviridis (anamorph Trichoderma atroviride), a screening system was designed that consists of a combination of Biolog Phenotype MicroArray plates, which contain 95 different carbon sources, and specific enzyme activity measurements using a chromogenic substrate. The results revealed growth-dependent kinetics of NAGase formation and it was shown that NAGase activities were enhanced on carbon sources sharing certain structural properties, especially on α-glucans (e.g. glycogen, dextrin and maltotriose) and oligosaccharides containing galactose. Enzyme activities were assessed in the wild-type and a H. atroviridis Δnag1 strain to investigate the influence of the two NAGases, Nag1 and Nag2, on total NAGase activity. Reduction of NAGase levels in the Δnag1 strain in comparison to the wild-type was strongly carbon-source and growth-phase dependent, indicating the distinct physiological roles of the two proteins. The transcript abundance of nag1 and nag2 was increased on carbon sources with elevated NAGase activity, indicating transcriptional regulation of these genes. The screening method for the identification of carbon sources that induce enzymes or a gene of interest, as presented in this paper, can be adapted for other purposes if appropriate enzyme or reporter assays are available.
-
-
-
Methylglyoxal detoxification by an aldo-keto reductase in the cyanobacterium Synechococcus sp. PCC 7002
More LessAldo-keto reductases (AKRs) are a superfamily of enzymes that reduce aldehydes and ketones, and have a broad range of substrates. An AKR gene, sakR1, was identified in the cyanobacterium Synechococcus sp. PCC 7002. A mutant strain with sakR1 inactivated was sensitive to glycerol, a carbon source that can support heterotrophic growth of Synechococcus sp. PCC 7002. It was found that the sakR1 null mutant accumulated more toxic methylglyoxal than the wild-type when glycerol was added to growth medium, suggesting that SakR1 is involved in the detoxification of methylglyoxal, a highly toxic metabolite that can damage cellular macromolecules. Enzymic analysis of recombinant SakR1 protein showed that it can efficiently reduce methylglyoxal with NADPH. Based on immunoblotting, SakR1 was not upregulated at an increased cellular methylglyoxal concentration. A pH-dependent enzyme-activity profile suggested that SakR1 activity could be regulated by cellular pH in Synechococcus sp. PCC 7002. The broad substrate specificity of SakR1 implies that SakR1 could play other roles in cellular metabolism.
-
-
-
Role of the Escherichia coli nitrate transport protein, NarU, in survival during severe nutrient starvation and slow growth
More LessEscherichia coli K-12 strains expressing either NarU or NarK as the only nitrate transport protein are both able to support nitrate-dependent anaerobic growth. The narK gene is highly expressed during anaerobic growth in the presence of nitrate, consistent with a role for NarK in nitrate transport coupled to nitrate reduction by the most active nitrate reductase encoded by the adjacent narGHJI operon. The physiological role of NarU is unknown. Reverse transcriptase PCR experiments established that, unlike the monocistronic narK gene, narU is co-transcribed with narZ as the first gene of a five-gene narUZYWV operon. The narK and narU genes were fused in-frame to a myc tag: the encoded fusion proteins complemented the nitrate-dependent growth defect of chromosomal narK and narU mutations. A commercial anti-Myc antibody was used to detect NarK and NarU in membrane fractions. During anaerobic growth in the presence of nitrate, the quantity of NarU-Myc accumulated during exponential growth was far less than that of NarK-Myc, but NarU was more abundant than NarK in stationary-phase cultures in the absence of nitrate. Although the concentration of NarU-Myc increased considerably during the post-exponential phase of growth, NarK-Myc was still more abundant than NarU-Myc in stationary-phase bacteria in the presence of nitrate. In chemostat competition experiments, a strain expressing only narU had a selective advantage relative to a strain expressing only narK during nutrient starvation or very slow growth, but NarK+ bacteria had a much greater selective advantage during rapid growth. The data suggest that NarU confers a selective advantage during severe nutrient starvation or slow growth, conditions similar to those encountered in vivo.
-
- Biodiversity And Evolution
-
-
-
Occurrence, phylogeny and evolution of ribulose-1,5-bisphosphate carboxylase/oxygenase genes in obligately chemolithoautotrophic sulfur-oxidizing bacteria of the genera Thiomicrospira and Thioalkalimicrobium
More LessThe occurrence of the different genes encoding ribulose-1,5-bisphosphate carboxylase/oxygenase (RubisCO), the key enzyme of the Calvin–Benson–Bassham cycle of autotrophic CO2 fixation, was investigated in the members of the genus Thiomicrospira and the relative genus Thioalkalimicrobium, all obligately chemolithoautotrophic sulfur-oxidizing Gammaproteobacteria. The cbbL gene encoding the ‘green-like’ form I RubisCO large subunit was found in all analysed species, while the cbbM gene encoding form II RubisCO was present only in Thiomicrospira species. Furthermore, species belonging to the Thiomicrospira crunogena 16S rRNA-based phylogenetic cluster also possessed two genes of green-like form I RubisCO, cbbL-1 and cbbL-2. Both 16S-rRNA- and cbbL-based phylogenies of the Thiomicrospira–Thioalkalimicrobium–Hydrogenovibrio group were congruent, thus supporting its monophyletic origin. On the other hand, it also supports the necessity for taxonomy reorganization of this group into a new family with four genera.
-
-
- Environmental Microbiology
-
-
-
Cloning and expression of the gene for periplasmic poly(vinyl alcohol) dehydrogenase from Sphingomonas sp. strain 113P3, a novel-type quinohaemoprotein alcohol dehydrogenase
More LessA gene for periplasmic poly(vinyl alcohol) (PVA) dehydrogenase (PVADH) was cloned, based on the N-terminal amino acid sequence of the purified PVADH from Sphingomonas sp. 113P3 and the sequence of the gene for PVADH (pvaA, GenBank accession no. AB190288). The recombinant PVADH tagged with hexahistidine was expressed in Escherichia coli and purified to homogeneity. The recombinant enzyme had the same characteristics as the purified enzyme from Sphingomonas sp. strain 113P. In addition to PVA, the recombinant PVADH could oxidize glycols such as polypropylene glycols and 1,3-butane/cyclohexanediol and 2,4-pentanediol, but neither primary nor secondary alcohols. The amino acid sequence of the recombinant PVADH showed similarity with those of PVADH from Pseudomonas sp. strain VM15C, putative PVADHs from Azoarcus sp. EbN1, and Xanthomonas species (54–25 % identity), and the quinohaemoprotein alcohol dehydrogenases (QH-ADHs) from Comamonas testosteroni, Ralstonia eutropha and Pseudomonas putida (25–29 % identity). PVADHs from strains 113P3 and VM15C have a conserved superbarrel domain (SD), probable PQQ-binding amino acids in the SD and a haem-binding domain (HBD) (they should be designated QH-PVADHs), but the positions of the amino acid sequences for the HBD and SD are the reverse of those of QH-ADHs. A protein structure of QH-PVADHs is proposed. Results of dot-blot hybridization and RT-PCR indicated that the three genes encoding oxidized PVA hydrolase, PVADH and cytochrome c are expressed constitutively and form an operon.
-
-
- Genes And Genomes
-
-
-
Two novel conjugative plasmids from a single strain of Sulfolobus
More LessTwo conjugative plasmids (CPs) were isolated and characterized from the same ‘Sulfolobus islandicus’ strain, SOG2/4. The plasmids were separated from each other and transferred into Sulfolobus solfataricus. One has a high copy number and is not stable (pSOG1) whereas the other has a low copy number and is stably maintained (pSOG2). Plasmid pSOG2 is the first Sulfolobus CP found to have these characteristics. The genomes of both pSOG plasmids have been sequenced and were compared to each other and the available Sulfolobus CPs. Interestingly, apart from a very well-conserved core, 70 % of the pSOG1 and pSOG2 genomes is largely different and composed of a mixture of genes that often resemble counterparts in previously described Sulfolobus CPs. However, about 20 % of the predicted genes do not have known homologues, not even in other CPs. Unlike pSOG1, pSOG2 does not contain a gene for the highly conserved PlrA protein nor for obvious homologues of partitioning proteins. Unlike pNOB8 and pKEF9, both pSOG plasmids lack the so-called clustered regularly interspaced short palindrome repeats (CRISPRs). The sites of recombination between the two genomes can be explained by the presence of recombination motifs previously identified in other Sulfolobus CPs. Like other Sulfolobus CPs, the pSOG plasmids possess a gene encoding an integrase of the tyrosine recombinase family. This integrase probably mediates plasmid site-specific integration into the host chromosome at the highly conserved tRNAGlu loci.
-
-
-
-
Horizontal transfer of the immunoglobulin A1 protease gene (iga) from Streptococcus to Gemella haemolysans
More LessBacterial IgA1 proteases share the ability to cleave human IgA1 at the hinge region. Nature has developed this trait along at least five independent evolutionary lineages. To obtain further insight into the phylogeny and function of IgA1 proteases, the nucleotide sequence of the iga gene that encodes the IgA1 protease was determined from two Streptococcus mitis strains and one Gemella haemolysans strain. Heterologous expression in Escherichia coli confirmed that the genes encode human IgA1-cleaving activity. IgA1 proteases from Streptococcus and G. haemolysans shared structural features, including a motif typical for zinc-dependent metalloproteases of clan MA(E) family M26 and an N-terminal signal sequence followed by an LPXTG cell-wall-anchor motif and two putative membrane-spanning domains. In addition, they all harboured a repeat region preceding the active site of the protease. In the streptococcal IgA1 proteases, a G5 domain, which has been suggested to bind N-acetylglucosamine, was identified. Conservation of these structures in otherwise diverse proteases suggests that they are essential to the biological function of the enzyme. The phylogenetic distribution of homologous iga genes and conservation of gene order in the iga gene region in different Streptococcus species, combined with the sequence homologies, strongly suggest that the iga gene is more ancient in Streptococcus than in G. haemolysans, and therefore that the IgA1 protease gene was transferred from Streptococcus to G. haemolysans.
-
- Pathogens And Pathogenicity
-
-
-
Disruption of the Aspergillus fumigatus ECM33 homologue results in rapid conidial germination, antifungal resistance and hypervirulence
More LessThe ECM33/SPS2 family of glycosylphosphatidylinositol-anchored proteins plays an important role in maintaining fungal cell wall integrity and virulence. However, the precise molecular role of these proteins is unknown. In this work, AfuEcm33, the gene encoding the ECM33 homologue in the important pathogenic fungus Aspergillus fumigatus, has been cloned and its function analysed. It is shown that disruption of AfuEcm33 results in rapid conidial germination, increased cell–cell adhesion, resistance to the antifungal agent caspofungin and increased virulence in an immunocompromised mouse model for disseminated aspergillosis. These results suggest that the protein encoded by AfuEcm33 is involved in key aspects of cell wall morphogenesis and plays an important role in A. fumigatus virulence.
-
-
-
-
Antigen-43-mediated autoaggregation impairs motility in Escherichia coli
More LessFunctional interaction between bacterial surface-displayed autoaggregation proteins such as antigen 43 (Ag43) of Escherichia coli and motility organelles such as flagella has not previously been described. Here, it has been demonstrated for the first time that Ag43-mediated aggregation can inhibit bacterial motility. Ag43 overexpression produces a dominant aggregation phenotype that overrides motility in the presence of low levels of flagella. In contrast, induction of an increased flagellation state prevents Ag43-mediated aggregation. This phenomenon was observed in naturally occurring subpopulations of E. coli as phase variants expressing and not expressing Ag43 revealed contrasting motility phenotypes. The effects were shown to be part of a general mechanism because other short adhesins capable of mediating autoaggregation (AIDA-I and TibA) also impaired motility. These novel insights into the function of bacterial autoaggregation proteins suggest that a balance between these two systems, i.e. autoaggregation and flagellation, influences motility.
-
-
-
Distribution of prophages and SGI-1 antibiotic-resistance genes among different Salmonella enterica serovar Typhimurium isolates
More LessRecently, the authors identified Salmonella enterica serovar Typhimurium (S. Typhimurium) definitive type (DT)104-specific sequences of mainly prophage origin by genomic subtractive hybridization. In the present study, the distribution of the prophages identified, ST104 and ST64B, and the novel prophage remnant designated prophage ST104B, was tested among 23 non-DT104 S. Typhimurium isolates of different phage types and 19 isolates of the DT104 subtypes DT104A, DT104B low and DT104L, and the DT104-related type U302. The four S. Typhimurium prophages Gifsy-1, Gifsy-2, Fels-1 and Fels-2 were also included. Analysis of prophage distribution in different S. Typhimurium isolates may supply additional information to enable development of a molecular method as an alternative to phage typing. Furthermore, the presence of the common DT104 antibiotic resistance genes for the penta-resistance type ACSSuT, aadA2, floR, pse-1, sul1 and tet(G), was also studied because of the authors' focus on this emerging type. Based on differences in prophage presence within their genome, it was possible to divide S. Typhimurium isolates into 12 groups. Although no clear relationship was found between different phage type and prophage presence, discrimination could be made between the different DT104 subtypes based on diversity in the presence of prophages ST104, ST104B and ST64B. The novel prophage remnant ST104B, which harbours a homologue of the Escherichia coli O157 : H7 HldD LPS assembly-related protein, was identified only in the 14 DT104L isolates and in the DT104-related U302 isolate. In conclusion, the presence of the genes for penta-resistance type ACSSuT, the HldD homologue containing ST104 prophage remnant and phage type DT104L are most likely common features of the emerging subtype of S. Typhimurium DT104.
-
-
-
Proteus mirabilis isolates of different origins do not show correlation with virulence attributes and can colonize the urinary tract of mice
More LessProteus mirabilis has been described as an aetiological agent in a wide range of infections, playing an important role in urinary tract infections (UTIs). In this study, a collection of P. mirabilis isolates obtained from clinical and non-clinical sources was analysed in order to determine a possible correlation between origin, virulence factors and in vivo infectivity. Isolates were characterized in vitro, assessing several virulence properties that had been previously associated with P. mirabilis uropathogenicity. Swarming motility, urease production, growth in urine, outer-membrane protein patterns, ability to grow in the presence of different iron sources, haemolysin and haemagglutinin production, and the presence and expression of diverse fimbrial genes, were analysed. In order to evaluate the infectivity of the different isolates, the experimental ascending UTI model in mice was used. Additionally, the Dienes test and the enterobacterial repetitive intergenic consensus (ERIC)-PCR assay were performed to assess the genetic diversity of the isolates. The results of the present study did not show any correlation between distribution of the diverse potential urovirulence factors and isolate source. No significant correlation was observed between infectivity and the origin of the isolates, since they all similarly colonized the urinary tract of the challenged mice. Finally, all isolates showed unique ERIC-PCR patterns, indicating that the isolates were genetically diverse. The results obtained in this study suggest that the source of P. mirabilis strains cannot be correlated with pathogenic attributes, and that the distribution of virulence factors between isolates of different origins may correspond to the opportunistic nature of the organism.
-
- Physiology
-
-
-
Osmotic stress in Synechocystis sp. PCC 6803: low tolerance towards nonionic osmotic stress results from lacking activation of glucosylglycerol accumulation
More LessIn order to compare the molecular principles of the acclimatization of bacterial cells to salt and nonionic osmotic stress, the moderately halotolerant cyanobacterium Synechocystis sp. PCC 6803 was challenged by salt (NaCl), and the osmolytes sorbitol and maltose. The physiological response towards each of the three compounds was found to be different. After salt addition, the cell volume remained unchanged, and the accumulation of the osmoprotective compound glucosylglycerol (GG) was observed after activation of the key enzyme GgpS at the biochemical and gene (ggpS) expression level. Sorbitol addition had only minor effects on the cell volume. In spite of the fact that the ggpS expression was increased, the GgpS enzyme was not activated, resulting in the absence of GG accumulation. In contrast the cells accumulated sorbitol, which served as a compatible solute and assured a certain osmotic resistance. In comparison to NaCl and sorbitol, the addition of maltose caused a strong decrease in cell volume indicating water efflux. However, no osmolyte accumulation was observed, resulting in an osmosensitive phenotype. Consequently, a successful response of Synechocystis cells to an osmotic challenge is indicative of the de novo synthesis of GG upon salt-dependent activation of the GgpS enzyme or the uptake of external solutes.
-
-
-
-
Candida albicans SNO1 and SNZ1 expressed in stationary-phase planktonic yeast cells and base of biofilm
More LessThe Candida albicans homologues of the most studied Saccharomyces cerevisiae stationary-phase genes, SNO1 and SNZ1, were used to test the hypothesis that, within a biofilm, some cells reach stationary phase within continuously fed, as well as static, C. albicans biofilms grown on dental acrylic. The authors first studied the expression patterns of these two genes in planktonic growth conditions. Using real-time RT-PCR (RT-RTPCR), increased peak expression of both SNZ1 and SNO1 was observed at 5 and 6 days, respectively, in C. albicans grown in suspension culture. SNZ1–yellow fluorescent protein (YFP) and SNO1–YFP were constructed to study expression at the cellular level and protein localization in C. albicans. Snz1p–YFP and Sno1p–YFP localized to the cytoplasm with maximum expression (>90 %) at 5 and 6 days, respectively, in planktonic conditions. When yeast growth was reinitiated, loss of fluorescence began immediately. Germ tubes and hyphae were non-fluorescent. Pseudohyphae began appearing at 9 days in planktonic yeast culture and expressed each protein by 11 days; however, the cells budding from pseudohyphae were not fluorescent. Biofilm was formed in vitro under either static or continuously fed conditions. Increased expression of the two genes was shown by RT-RTPCR, beginning by day 3 and increasing through to day 15 (continuously fed biofilm). Only the bottommost layer of acrylic-adhered cells in the biofilm showed 25 and 40 % fluorescence at 6 and 15 days, respectively. These observations suggest that only a few cells in C. albicans biofilms express genes associated with the planktonic stationary phase and that these are found at the bottom of the biofilm adhered to the surface.
-
-
-
Effect of AmtB homologues on the post-translational regulation of nitrogenase activity in response to ammonium and energy signals in Rhodospirillum rubrum
More LessThe AmtB protein transports uncharged NH3 into the cell, but it also interacts with the nitrogen regulatory protein PII, which in turn regulates a variety of proteins involved in nitrogen fixation and utilization. Three PII homologues, GlnB, GlnK and GlnJ, have been identified in the photosynthetic bacterium Rhodospirillum rubrum, and they have roles in at least four overlapping and distinct functions, one of which is the post-translational regulation of nitrogenase activity. In R. rubrum, nitrogenase activity is tightly regulated in response to
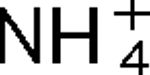 addition or energy depletion (shift to darkness), and this regulation is catalysed by the post-translational regulatory system encoded by draTG. Two amtB homologues, amtB
1 and amtB
2, have been identified in R. rubrum, and they are linked with glnJ and glnK, respectively. Mutants lacking AmtB1 are defective in their response to both
addition or energy depletion (shift to darkness), and this regulation is catalysed by the post-translational regulatory system encoded by draTG. Two amtB homologues, amtB
1 and amtB
2, have been identified in R. rubrum, and they are linked with glnJ and glnK, respectively. Mutants lacking AmtB1 are defective in their response to both  addition and darkness, while mutants lacking AmtB2 show little effect on the regulation of nitrogenase activity. These responses to darkness and
addition and darkness, while mutants lacking AmtB2 show little effect on the regulation of nitrogenase activity. These responses to darkness and 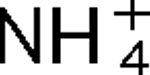 appear to involve different signal transduction pathways, and the poor response to darkness does not seem to be an indirect result of perturbation of internal pools of nitrogen. It is also shown that AmtB1 is necessary to sequester detectable amounts GlnJ to the cell membrane. These results suggest that some element of the AmtB1-PII regulatory system senses energy deprivation and a consistent model for the integration of nitrogen, carbon and energy signals by PII is proposed. Other results demonstrate a degree of specificity in interaction of AmtB1 with the different PII homologues in R. rubrum. Such interaction specificity might be important in explaining the way in which PII proteins regulate processes involved in nitrogen acquisition and utilization.
appear to involve different signal transduction pathways, and the poor response to darkness does not seem to be an indirect result of perturbation of internal pools of nitrogen. It is also shown that AmtB1 is necessary to sequester detectable amounts GlnJ to the cell membrane. These results suggest that some element of the AmtB1-PII regulatory system senses energy deprivation and a consistent model for the integration of nitrogen, carbon and energy signals by PII is proposed. Other results demonstrate a degree of specificity in interaction of AmtB1 with the different PII homologues in R. rubrum. Such interaction specificity might be important in explaining the way in which PII proteins regulate processes involved in nitrogen acquisition and utilization.
-
-
-
Global gene expression in Escherichia coli K-12 during short-term and long-term adaptation to glucose-limited continuous culture conditions
More LessMicroarray technology was used to study the cellular events that take place at the transcription level during short-term (physiological) and long-term (genetic) adaptation of the faecal indicator bacterium Escherichia coli K-12 to slow growth under limited nutrient supply. Short-term and long-term adaptation were assessed by comparing the mRNA levels isolated after 40 or 500 h of glucose-limited continuous culture at a dilution rate of 0.3 h−1 with those from batch culture with glucose excess. A large number of genes encoding periplasmic binding proteins were upregulated, indicating that the cells are prepared for high-affinity uptake of all types of carbon sources during glucose-limited growth in continuous culture. All the genes belonging to the maltose (mal/lamB) and galactose (mgl/gal) operons were upregulated. A similar transcription pattern was observed for long-term cultures except that the expression factors were lower than in the short-term adaptation. The patterns of upregulation were confirmed by real-time RT-PCR. A switch from a fully operational citric acid cycle to the PEP-glyoxylate cycle was clearly observed in cells grown in glucose-limited continuous culture when compared to batch-grown cells and this was confirmed by transcriptome analysis. This transcriptome analysis confirms and extends the observations from previous proteome and catabolome studies in the authors' laboratory.
-
-
-
Functional identification of ygiP as a positive regulator of the ttdA-ttdB-ygjE operon
More LessFunctional characterization of unknown genes is currently a major task in biology. The search for gene function involves a combination of various in silico, in vitro and in vivo approaches. Available knowledge from the study of more than 21 LysR-type regulators in Escherichia coli has facilitated the classification of new members of the family. From sequence similarities and its location on the E. coli chromosome, it is suggested that ygiP encodes a lysR regulator controlling the expression of a neighbouring operon; this operon encodes the two subunits of tartrate dehydratase (TtdA, TtdB) and YgiE, an integral inner-membrane protein possibly involved in tartrate uptake. Expression of tartrate dehydratase, which converts tartrate to oxaloacetate, is required for anaerobic growth on glycerol as carbon source in the presence of tartrate. Here, it has been demonstrated that disruption of ygiP, ttdA or ygjE abolishes tartrate-dependent anaerobic growth on glycerol. It has also been shown that tartrate-dependent induction of the ttdA-ttdB-ygjE operon requires a functional YgiP.
-
- Plant-Microbe Interactions
-
-
-
Antimicrobial activity of potato aspartic proteases (StAPs) involves membrane permeabilization
More LessSolanum tuberosum aspartic proteases (StAPs) with antimicrobial activity are induced after abiotic and biotic stress. In this study the ability of StAPs to produce a direct antimicrobial effect was investigated. Viability assays demonstrated that StAPs are able to kill spores of Fusarium solani and Phytophthora infestans in a dose-dependent manner. Localization experiments with FITC-labelled StAPs proved that the proteins interact directly with the surface of spores and hyphae of F. solani and P. infestans. Moreover, incubation of spores and hyphae with StAPs resulted in membrane permeabilization, as shown by the uptake of the fluorescent dye SYTOX Green. It is concluded that the antimicrobial effect of StAPs against F. solani and P. infestans is caused by a direct interaction with the microbial surfaces followed by membrane permeabilization.
-
-
-
-
Isolation of salt-sensitive mutants of Sinorhizobium meliloti strain Rm1021
More LessThe determinants necessary for adaptation to high NaCl concentrations and competition for nodule occupancy in Sinorhizobium meliloti were investigated genetically. Mutations in fabG as well as smc02909 (transmembrane transglycosylase), trigger factor (tig) and smc00717 (probably ftsE) gave rise to strains that were unable to tolerate high salt and were uncompetitive for nodule occupancy relative to the wild-type. Moreover exoF1, exoA and pgm determinants were determined to be necessary for strain Rm1021 to survive high NaCl and/or MgCl2 concentrations. The introduction of an expR + allele was capable of suppressing the Mg2+ sensitivity associated with the exoF1, but not the exoA, mutation in a manner independent of exopolysaccharide II (EPS II)-associated mucoidy. The results also show that the EPS II-associated mucoid phenotype was affected by either Mg2+or K+, but not by Li+, Ca2+, or high osmolarity.
-
-
-
Characterization of genes involved in erythritol catabolism in Rhizobium leguminosarum bv. viciae
More LessA genetic locus encoding erythritol uptake and catabolism genes was identified in Rhizobium leguminosarum bv. viciae, and shown to be plasmid encoded in a wide range of R. leguminosarum strains. A Tn5-B22 mutant (19B-3) unable to grow on erythritol was isolated from a mutant library of R. leguminosarum strain VF39SM. The mutated gene eryF was cloned and partially sequenced, and determined to have a high homology to permease genes of ABC transporters. A cosmid complementing the mutation (pCos42) was identified and was shown to carry all the genes necessary to restore the ability to grow on erythritol to a VF39SM strain cured of pRleVF39f. In the genomic DNA sequence of strain 3841, the gene linked to the mutation in 19B-3 is flanked by a cluster of genes with high homology to the known erythritol catabolic genes from Brucella spp. Through mutagenesis studies, three distinct operons on pCos42 that are required for growth on erythritol were identified: an ABC-transporter operon (eryEFG), a catabolic operon (eryABCD) and an operon (deoR-tpiA2-rpiB) that encodes a gene with significant homology to triosephosphate isomerase (tpiA2). These genes all share high sequence identity to genes in the erythritol catabolism region of Brucella spp., and clustalw alignments suggest that horizontal transfer of the erythritol locus may have occurred between R. leguminosarum and Brucella. Transcription of the eryABCD operon is repressed by EryD and is induced by the presence of erythritol. Mutant 19B-3 was impaired in its ability to compete against wild-type for nodulation of pea plants but was still capable of forming nitrogen-fixing nodules.
-
Volumes and issues
-
Volume 170 (2024)
-
Volume 169 (2023)
-
Volume 168 (2022)
-
Volume 167 (2021)
-
Volume 166 (2020)
-
Volume 165 (2019)
-
Volume 164 (2018)
-
Volume 163 (2017)
-
Volume 162 (2016)
-
Volume 161 (2015)
-
Volume 160 (2014)
-
Volume 159 (2013)
-
Volume 158 (2012)
-
Volume 157 (2011)
-
Volume 156 (2010)
-
Volume 155 (2009)
-
Volume 154 (2008)
-
Volume 153 (2007)
-
Volume 152 (2006)
-
Volume 151 (2005)
-
Volume 150 (2004)
-
Volume 149 (2003)
-
Volume 148 (2002)
-
Volume 147 (2001)
-
Volume 146 (2000)
-
Volume 145 (1999)
-
Volume 144 (1998)
-
Volume 143 (1997)
-
Volume 142 (1996)
-
Volume 141 (1995)
-
Volume 140 (1994)
-
Volume 139 (1993)
-
Volume 138 (1992)
-
Volume 137 (1991)
-
Volume 136 (1990)
-
Volume 135 (1989)
-
Volume 134 (1988)
-
Volume 133 (1987)
-
Volume 132 (1986)
-
Volume 131 (1985)
-
Volume 130 (1984)
-
Volume 129 (1983)
-
Volume 128 (1982)
-
Volume 127 (1981)
-
Volume 126 (1981)
-
Volume 125 (1981)
-
Volume 124 (1981)
-
Volume 123 (1981)
-
Volume 122 (1981)
-
Volume 121 (1980)
-
Volume 120 (1980)
-
Volume 119 (1980)
-
Volume 118 (1980)
-
Volume 117 (1980)
-
Volume 116 (1980)
-
Volume 115 (1979)
-
Volume 114 (1979)
-
Volume 113 (1979)
-
Volume 112 (1979)
-
Volume 111 (1979)
-
Volume 110 (1979)
-
Volume 109 (1978)
-
Volume 108 (1978)
-
Volume 107 (1978)
-
Volume 106 (1978)
-
Volume 105 (1978)
-
Volume 104 (1978)
-
Volume 103 (1977)
-
Volume 102 (1977)
-
Volume 101 (1977)
-
Volume 100 (1977)
-
Volume 99 (1977)
-
Volume 98 (1977)
-
Volume 97 (1976)
-
Volume 96 (1976)
-
Volume 95 (1976)
-
Volume 94 (1976)
-
Volume 93 (1976)
-
Volume 92 (1976)
-
Volume 91 (1975)
-
Volume 90 (1975)
-
Volume 89 (1975)
-
Volume 88 (1975)
-
Volume 87 (1975)
-
Volume 86 (1975)
-
Volume 85 (1974)
-
Volume 84 (1974)
-
Volume 83 (1974)
-
Volume 82 (1974)
-
Volume 81 (1974)
-
Volume 80 (1974)
-
Volume 79 (1973)
-
Volume 78 (1973)
-
Volume 77 (1973)
-
Volume 76 (1973)
-
Volume 75 (1973)
-
Volume 74 (1973)
-
Volume 73 (1972)
-
Volume 72 (1972)
-
Volume 71 (1972)
-
Volume 70 (1972)
-
Volume 69 (1971)
-
Volume 68 (1971)
-
Volume 67 (1971)
-
Volume 66 (1971)
-
Volume 65 (1971)
-
Volume 64 (1970)
-
Volume 63 (1970)
-
Volume 62 (1970)
-
Volume 61 (1970)
-
Volume 60 (1970)
-
Volume 59 (1969)
-
Volume 58 (1969)
-
Volume 57 (1969)
-
Volume 56 (1969)
-
Volume 55 (1969)
-
Volume 54 (1968)
-
Volume 53 (1968)
-
Volume 52 (1968)
-
Volume 51 (1968)
-
Volume 50 (1968)
-
Volume 49 (1967)
-
Volume 48 (1967)
-
Volume 47 (1967)
-
Volume 46 (1967)
-
Volume 45 (1966)
-
Volume 44 (1966)
-
Volume 43 (1966)
-
Volume 42 (1966)
-
Volume 41 (1965)
-
Volume 40 (1965)
-
Volume 39 (1965)
-
Volume 38 (1965)
-
Volume 37 (1964)
-
Volume 36 (1964)
-
Volume 35 (1964)
-
Volume 34 (1964)
-
Volume 33 (1963)
-
Volume 32 (1963)
-
Volume 31 (1963)
-
Volume 30 (1963)
-
Volume 29 (1962)
-
Volume 28 (1962)
-
Volume 27 (1962)
-
Volume 26 (1961)
-
Volume 25 (1961)
-
Volume 24 (1961)
-
Volume 23 (1960)
-
Volume 22 (1960)
-
Volume 21 (1959)
-
Volume 20 (1959)
-
Volume 19 (1958)
-
Volume 18 (1958)
-
Volume 17 (1957)
-
Volume 16 (1957)
-
Volume 15 (1956)
-
Volume 14 (1956)
-
Volume 13 (1955)
-
Volume 12 (1955)
-
Volume 11 (1954)
-
Volume 10 (1954)
-
Volume 9 (1953)
-
Volume 8 (1953)
-
Volume 7 (1952)
-
Volume 6 (1952)
-
Volume 5 (1951)
-
Volume 4 (1950)
-
Volume 3 (1949)
-
Volume 2 (1948)
-
Volume 1 (1947)
Most Read This Month
