 -
Volume 153,
Issue 7,
2007
-
Volume 153,
Issue 7,
2007

Volume 153, Issue 7, 2007
- Cell And Developmental Biology
-
-
-
The MAP kinase TVK1 regulates conidiation, hydrophobicity and the expression of genes encoding cell wall proteins in the fungus Trichoderma virens
More LessMitogen-activated protein (MAP) kinases modulate morphological and genetic processes, which determine cell fate. The tvk1 gene encodes a MAP kinase of Trichoderma virens and its deletion promotes an unusual conidiation phenotype in submerged culture. Here, it is reported that the morphology, physiology and expression of genes encoding cell wall proteins from Trichoderma are significantly affected by Tvk1. Morphological changes were evident in the cell walls of aerial conidia produced by a MAPK null mutant when compared to those produced by the wild-type. Unexpectedly, conidia produced in submerged culture by the Δtvk1 strain were highly hydrophobic, whereas in aerial conidia hydrophobicity was severely reduced. In addition, the Δtvk1 strain was unable to break the liquid–air interface when the fungus grew in rich medium; however, when it grew in minimal medium the fungus produced large filaments which were much more efficient at breaking the interface than the wild-type. Through cDNA subtractive hybridization between the wild-type and Δtvk1 grown in submerged culture, five genes encoding hydrophobin-like proteins and two additional genes encoding cell wall proteins were identified. Four hydrophobin-encoding genes (Tv-hfb1, Tv-srh1, tv-cfth1 and Tv-qid3) and a gene encoding a clock-controlled-like protein (Tv-ccg14/TvSm1) were upregulated in Δtvk1, whereas genes encoding a cell wall protein (tv-qid74) and an additional hydrophobin (tv-hfb3) were absent in the mutant strain. Clear differences in gene expression were shown during conidiation and emergence from the liquid–air interface, suggesting different functions of the corresponding proteins in these two phenomena. The results support a model in which Tvk1 regulates morphology and genes encoding cell wall proteins during development of Trichoderma.
-
-
- Biochemistry And Molecular Biology
-
-
-
Malic enzyme: the controlling activity for lipid production? Overexpression of malic enzyme in Mucor circinelloides leads to a 2.5-fold increase in lipid accumulation
More LessMalic enzyme (ME; NADP+-dependent; EC 1 . 1 . 1 . 40) has been postulated to be the rate-limiting step for fatty acid biosynthesis in oleaginous fungi in which the extent of lipid accumulation is below the maximum possible. The genes encoding the isoform of ME involved in fatty acid synthesis were identified in Mucor circinelloides and Mortierella alpina, two commercially useful oil-producing fungi, using degenerate primers. Both showed high similarity with ME genes from other micro-organisms. The whole-length ME gene from each source was cloned into a leucine auxotroph of Mc. circinelloides and placed under the control of the constitutive glyceraldehyde-3-phosphate dehydrogenase gene (gpd1) promoter. After confirming correct expression of the ME genes, the two recombinant strains were grown in fully controlled, submerged-culture bioreactors using a high C : N ratio medium for lipid accumulation. Activities of ME were increased by two- to threefold and the lipid contents of the cells, in both recombinants, were increased from 12 % of the biomass to 30 %. Simultaneously, the degree of fatty acid desaturation increased slightly. Thus, increased expression of the ME gene leads to both increased biosynthesis of fatty acids and formation of unsaturated fatty acids, including γ-linolenic acid (18 : 3 n-6). At the end of lipid accumulation (96 h), ME activity in the recombinant strains had ceased, as it had done in the parent wild-type cells, indicating that additional, but unknown, controls over its activity must be in place to account for this loss of activity: this may be due to the presence of a specific ME-cleaving enzyme. The hypothesis that the rate-limiting step of fatty acid biosynthesis is therefore the supply of NADPH, as generated specifically and solely by ME, is therefore considerably strengthened by these results.
-
-
-
-
A novel type of subtilase from the psychrotolerant bacterium Pseudoalteromonas sp. SM9913: catalytic and structural properties of deseasin MCP-01
More LessMCP-01, the main protease secreted by the deep-sea cold-adapted bacterium Pseudoalteromonas sp. SM9913, is a cold-adapted serine protease. Gene mcp01 encoding MCP-01 contains an ORF of 2508 bp encoding a protein of 835 amino acid residues with an M r of 87 773 Da, which is a multidomain subtilase precursor. Mature MCP-01 purified from the culture of strain SM9913 with an M r of 65.84 kDa is a multidomain protein composed of a catalytic domain, a linker, a P_proprotein domain and a polycystic kidney disease (PKD) domain. To the best of the authors' knowledge, no mature subtilase has been reported to date with this domain architecture. Phylogenetic analyses of subtilases showed that MCP-01 and 12 hypothetical proteins retrieved from public databases form a strongly supported group within the subtilase subfamily. These 13 proteins are predicted to share a similar domain architecture and represent a structurally novel group within the S8A subfamily. The substrate specificities of MCP-01 towards synthetic peptides differed from that of a typical S8A protease, subtilisin Carlsberg. Since most of this new subgroup of subtilases, including MCP-01 and the 12 MCP-01-like subtilases, are from deep-sea bacteria, they are termed deseasins. MCP-01 is the type example of a deseasin, since it is the only one that has been purified and characterized. In addition, the structural characteristics and catalytic properties of deseasin MCP-01 show that structurally and kinetically it is adapted to low temperatures.
-
-
-
The density of negative charge in the cell wall influences two-component signal transduction in Bacillus subtilis
More LessThe Dlt system modulates the density of negative charge in the cell wall of Gram-positive bacteria by substituting anionic polymers (wall and lipoteichoic acids) with d-alanine. The htrA and htrB genes, regulated by the CssRS two-component system (TCS) and encoding membrane-associated protein quality control proteases, were expressed at strongly decreased levels in a mutant with defective Dlt (dltD : : miniTn10) as compared to the dlt + wild-type strain under a secretion stress condition (hypersecretion of AmyQ α-amylase). The level of HtrA protein in the extracellular proteome of the dltD mutant was decreased consistently. Expression from the promoter of the liaIHGFSR (yvqIHGFEC) operon (P liaI ) is dependent on the LiaRS TCS. The Dlt defect increased the expression from P liaI under two stress conditions, AmyQ hypersecretion and treatment with a cationic antimicrobial peptide (LL-37), but decreased the expression in vancomycin-treated cells. Furthermore, Dlt inactivation enhanced the expression of the YxdJK-regulated yxdL gene in LL-37-treated cells. The increased net negative charge of the cell wall seems to cause varied and opposite effects on the expression of CssRS-, LiaRS- and YxdJK-regulated genes under different stress conditions. The results suggest that TCSs which sense misfolded proteins or peptides are modulated by the density of negative charge in the cell wall. The density of negative charge on the outer surface of the cell membrane did not have a similar effect on TCSs.
-
-
-
A group of Escherichia coli and Salmonella enterica O antigens sharing a common backbone structure
More LessThe O-antigen moiety of the LPS is one of the most variable cell surface components of the Gram-negative bacterial outer membrane. Variation is due to the presence of different sugars and sugar linkages. Here, it is reported that a group of Escherichia coli O serogroups (O17, O44, O73, O77 and O106), and the Salmonella enterica serogroup O : 6,14 (H), share a common four-sugar backbone O-subunit structure, and possess almost identical O-antigen gene clusters. Whereas the E. coli O77 antigen does not have any substitutions, the other O antigens in this group differ by the addition of one or two glucose side branches at various positions of the backbone. The O-antigen gene clusters for all members of the group encode only the proteins required for biosynthesis of the common four-sugar backbone. The identification of three genes within a putative prophage in the E. coli O44 genome is also reported; these genes are presumably involved in the glucosylation of the basic tetrasaccharide unit. This was confirmed by deletion of one of the genes, which encodes a putative glucosyltransferase. Structural analysis of the O antigen produced by the mutant strain demonstrated the absence of glucosylation. An O-antigen structure shared by five E. coli and one S. enterica serogroups, all of which have a long evolutionary history, suggests that the common backbone may be important for the survival of E. coli strains in the environment, or for their pathogenicity.
-
-
-
Three putative cation/proton antiporters from the soda lake alkaliphile Alkalimonas amylolytica N10 complement an alkali-sensitive Escherichia coli mutant
More LessAttempts to identify members of the antiporter complement of the alkali- and saline-adapted soda lake alkaliphile Alkalimonas amylolytica N10 have used screens of DNA libraries in antiporter-deficient Escherichia coli KNabc. Earlier screens used Na+ or Li+ for selection but only identified one NhaD-type antiporter whose properties were inconsistent with a robust role in pH homeostasis. Here, new screens using elevated pH for selection identified three other putative antiporter genes that conferred resistance to pH ≥8.5 as well as Na+ resistance. The three predicted gene products were in the calcium/cation antiporter (CaCA), cation/proton antiporter-2 (CPA2) and cation/proton antiporter-1 (CPA1) families of membrane transporters, and were designated Aa-CaxA, Aa-KefB and Aa-NhaP respectively, reflecting homology within those families. Aa-CaxA conferred the poorest Na+ resistance and also conferred modest Ca2+ resistance. Aa-KefB and Aa-NhaP inhibited growth of a K+ uptake-deficient E. coli mutant (TK2420), suggesting that they catalysed K+ efflux. For Aa-NhaP, the reversibility of the growth inhibition by high K+ concentrations depended upon an organic nitrogen source, e.g. glutamine, rather than ammonium. This suggests that
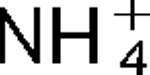 as well as K+ efflux is catalysed by Aa-NhaP. Vesicles of E. coli KNabc expressing Aa-NhaP, which conferred the strongest alkali resistance, exhibited K+/H+ antiport activity in a pH range from 7.5 to 9.5, and with an apparent K
m for K+ of 0.5 mM at pH 8.0. The properties of this antiporter are consistent with the possibility that this soda lake alkaliphile uses K+(
as well as K+ efflux is catalysed by Aa-NhaP. Vesicles of E. coli KNabc expressing Aa-NhaP, which conferred the strongest alkali resistance, exhibited K+/H+ antiport activity in a pH range from 7.5 to 9.5, and with an apparent K
m for K+ of 0.5 mM at pH 8.0. The properties of this antiporter are consistent with the possibility that this soda lake alkaliphile uses K+(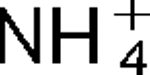 )/H+ antiport as part of its alkaline pH homeostasis mechanism and part of its capacity to reduce potentially toxic accumulation of cytoplasmic K+ or
)/H+ antiport as part of its alkaline pH homeostasis mechanism and part of its capacity to reduce potentially toxic accumulation of cytoplasmic K+ or 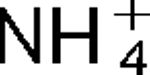 respectively, under conditions of high osmolarity or active amino acid catabolism.
respectively, under conditions of high osmolarity or active amino acid catabolism.
-
-
-
Expression of Corynebacterium glutamicum glycolytic genes varies with carbon source and growth phase
More LessA basic pattern of gene expression and of relative expression levels during different growth phases was obtained for Corynebacterium glutamicum R grown on different carbon sources. The gapA-pgk-tpi-ppc gene cluster was transcribed as a mono- or polycistronic mRNA, depending on the growth phase. The 1.4 kb (gapA) and 2.3 kb (pgk-tip) mRNAs were expressed in the early through late exponential phases, whereas the 3.7 kb (gapA-pgk-tpi) and 5.4 kb (pgk-tpi-ppc) mRNAs were only detected in the mid-exponential phase. All other glycolytic genes except pps, glk and pgi were transcribed as monocistronic mRNAs under all tested conditions. Identification and alignment of the promoter regions of the transcriptional start sites of glycolytic genes revealed strong similarities to the σ A consensus promoter sequences of Gram-positive bacteria. All genes involved in glycolysis were coordinately expressed in medium containing glucose. Growth in the presence of glucose gave rise to abundant expression of most glycolytic genes, with the level of gapA transcript being the highest. Glucose depletion led to a rapid repression of most glycolytic genes and a corresponding two- to fivefold increased expression of the gluconeogenic genes pps, pck and malE, which are induced by pyruvate, lactate, acetate and/or other organic acids.
-
-
-
Agmatine deiminase pathway genes in Lactobacillus brevis are linked to the tyrosine decarboxylation operon in a putative acid resistance locus
More LessIn lactic acid bacteria (LAB), amino acids and their derivatives may be converted into amine-containing compounds designated biogenic amines, in pathways providing metabolic energy and/or acid resistance to the bacteria. In a previous study, a pathway converting tyrosine to tyramine was detected in Lactobacillus brevis and a fragment of a gene possibly involved in the production of another biogenic amine, putrescine, from agmatine, was detected in the same locus. The present study was carried out to determine if Lb. brevis actually harbours two biogenic amine-producing pathways in the same locus and to investigate the occurrence of the two gene clusters in other bacteria. Sequencing of the DNA locus in Lb. brevis revealed a cluster of six genes that are related to previously reported genes of agmatine deiminase pathways but with marked differences such as two genes encoding putative agmatine deiminases rather than one. Heterologous expression of encoded enzymes confirmed the presence of at least one active agmatine deiminase and one amino acid transporter that efficiently exchanged agmatine and putrescine. It was concluded that the Lb. brevis gene cluster encodes a functional and highly specific agmatine deiminase pathway. Screening of a collection of 197 LAB disclosed the same genes in 36 strains from six different species, and almost all the positive bacteria also contained the tyrosine catabolic pathway genes in the same locus. These results support the hypothesis that the agmatine deiminase and tyrosine catabolic pathways belong to a genomic region that provides acid resistance and that is exchanged horizontally as a whole between LAB.
-
-
-
Ribulose bisphosphate carboxylase activity and a Calvin cycle gene cluster in Sulfobacillus species
More LessThe Calvin–Benson–Bassham (CBB) cycle has been extensively studied in proteobacteria, cyanobacteria, algae and plants, but hardly at all in Gram-positive bacteria. Some characteristics of ribulose bisphosphate carboxylase/oxygenase (RuBisCO) and a cluster of potential CBB cycle genes in a Gram-positive bacterium are described in this study with two species of Sulfobacillus (Gram-positive, facultatively autotrophic, mineral sulfide-oxidizing acidophiles). In contrast to the Gram-negative, iron-oxidizing acidophile Acidithiobacillus ferrooxidans, Sulfobacillus thermosulfidooxidans grew poorly autotrophically unless the CO2 concentration was enhanced over that in air. However, the RuBisCO of each organism showed similar affinities for CO2 and for ribulose 1,5-bisphosphate, and similar apparent derepression of activity under CO2 limitation. The red-type, form I RuBisCO of Sulfobacillus acidophilus was confirmed as closely related to that of the anoxygenic phototroph Oscillochloris trichoides. Eight genes potentially involved in the CBB cycle in S. acidophilus were clustered in the order cbbA, cbbP, cbbE, cbbL, cbbS, cbbX, cbbG and cbbT.
-
-
-
Involvement of a novel copper chaperone in tyrosinase activity and melanin synthesis in Marinomonas mediterranea
More LessTyrosinase activity and melanin synthesis in the marine bacterium Marinomonas mediterranea in media with very low copper concentrations are dependent on the presence of a protein (PpoB2) that functions as a chaperone to deliver copper to tyrosinase (PpoB1). Under these conditions, mutants in ppoB2 (such as strain T105) produce PpoB1 as an apoenzyme that can be reconstituted to the active holoenzyme by the addition of cupric ions to cell extracts. To study PpoB2 functionality, a system was developed for genetic complementation in M. mediterranea. Using this approach, melanin synthesis was restored in strain T105 when a wild-type copy of ppoB2 was introduced. PpoB2 is a novel protein since it is believed to be the first to be described that contains several motifs similar to metal binding motifs present separately in other types of copper-related protein. At least three motifs, a His-rich N-terminal region, and the short CxxxC and MxxxMM sequences, are essential for the functionality of PpoB2, since site-directed mutagenesis of these motifs resulted in a non-functional protein. In addition, it was demonstrated that PpoB2 is a membrane copper transporter putatively participating in the delivery of this ion specifically to the tyrosinase of M. mediterranea and not to a second copper oxidase showing laccase activity that this micro-organism also expresses. PpoB2 has similarities with the COG5486 group encoding putative transmembrane metal binding proteins, and is believed to be the first protein in this group to be experimentally characterized. It may constitute the first example of a novel type of protein involved in copper trafficking in bacteria.
-
-
-
Irreversible loss of membrane-binding activity of Listeria-derived cytolysins in non-acidic conditions: a distinct difference from allied cytolysins produced by other Gram-positive bacteria
More LessListeriolysin O (LLO), a member of the cholesterol-dependent cytolysin (CDC) family, is a major virulence factor of Listeria monocytogenes and contributes to bacterial escape from intracellular killing of macrophages. LLO is activated under weakly acidic conditions; however, the molecular mechanism of this pH-dependent expression of cytolytic activity of LLO is poorly understood. In this study, CDCs including LLO, ivanolysin O (ILO), seeligeriolysin O (LSO), pneumolysin (PLY), streptolysin O (SLO) and perfringolysin O (PFO) were prepared as recombinant proteins and examined for their functional changes after treatment under various pH conditions. Haemolytic and membrane cholesterol-binding activities were not affected in PLY, SLO and PFO at any pH examined. By contrast, all the Listeria-derived cytolysins, LLO, ILO and LSO, were active only at an acidic pH and rapidly inactivated under neutral or alkaline conditions. Once inactivated, LLO could not be reactivated even by a downward pH shift. The hydrophobicity of LLO treated at neutral or alkaline pH was increased. These data suggested that the pH-dependent loss of cytolytic activity appeared to be due to irreversible structural changes of domain 4 that resulted in the loss of target membrane cholesterol binding.
-
-
-
Deletions of recBCD or recD influence genetic transformation differently and are lethal together with a recJ deletion in Acinetobacter baylyi
More LessIn prokaryotes, homologous recombination is essential for the repair of genomic DNA damage and for the integration of DNA taken up during horizontal gene transfer. In Escherichia coli, the exonucleases RecJ (specific for 5′ single-stranded DNA) and RecBCD (degrades duplex DNA) play important roles in recombination and recombinational double-strand break (DSB) repair by the RecF and RecBCD pathways, respectively. The cloned recJ of Acinetobacter baylyi partially complemented an E. coli recJ mutant, suggesting functional similarity of the enzymes. A ΔrecJ mutant of A. baylyi was only slightly altered in transformability and was not affected in UV survival. In contrast, a ΔrecBCD mutant was UV-sensitive, and had a low viability and altered transformation. Compared to wild-type, transformation with large chromosomal DNA fragments was decreased about 5-fold, while transformation with 1.5 kbp DNA fragments was increased 3.3- to 7-fold. A ΔrecD mutation did not affect transformation, viability or UV resistance. However, double mutants recJ recBCD and recJ recD were non-viable, suggesting that the RecJ DNase or the RecBCD DNase (presumably absent in recD) becomes essential for the recombinational repair of spontaneously inactivated replication forks if the other DNase is absent. A model of recombination during genetic transformation is discussed in which the two ends of the single-stranded donor DNA present in the cytoplasm frequently integrate separately and often with a time difference. If replication runs through that genomic region before both ends of the donor DNA are ligated to recipient DNA, a double-strand break (DSB) is formed. In these cases, transformation becomes dependent on DSB repair.
-
-
-
Effect of proteasome inhibitor clasto-lactacystin-β-lactone on the proteome of the haloarchaeon Haloferax volcanii
More LessProteasomes play key roles in a variety of eukaryotic cell functions, including translation, transcription, metabolism, DNA repair and cell-cycle control. The biological functions of these multicatalytic proteases in archaea, however, are poorly understood. In this study, Haloferax volcanii was used as a model to determine the influence the proteasome-specific inhibitor clasto-lactacystin-β-lactone (cLβL) has on archaeal proteome composition. Addition of 20–30 μM cLβL had a widespread effect on the proteome, with a 38–42 % increase in the number of 2-D gel electrophoresis (2-DE) protein spots, from an average of 627 to 1036 spots. Protein identities for 17 of the spots that were easily separated by 2-DE and unique and/or increased 2- to 14-fold in the cLβL-treated cells were determined by tandem mass spectrometry (MS/MS). These included protein homologues of the DJ-1/ThiJ family, mobilization of sulfur system, translation elongation factor EF-1 A, ribosomal proteins, tubulin-like FtsZ, divalent metal ABC transporter, dihydroxyacetone kinase DhaL, aldehyde dehydrogenase and 2-oxoacid decarboxylase E1β. Based on these results, inhibition of H. volcanii proteasomes had a global influence on proteome composition, including proteins involved in central functions of the cell.
-
- Biodiversity And Evolution
-
-
-
Single nucleotide polymorphisms in the ectomycorrhizal mushroom Tricholoma matsutake
More LessSingle nucleotide polymorphisms (SNPs) are becoming increasingly popular markers for studying a variety of biological phenomena. This paper describes the development and analysis of a set of SNP markers for the basidiomycete fungus Tricholoma matsutake. T. matsutake is a gourmet mycorrhizal mushroom primarily associated with pine forests. However, little is known about its genetics and genomic variation, including SNP variation. To identify and analyse SNPs in T. matsutake, a genomic library was constructed and >72 000 nt were analysed from >200 random clones. Primers from 20 sequenced fragments were then designed to amplify and sequence >10 000 bp sequences from the original strain, from which the genomic library was constructed, as well as another strain from >350 km away; both strains were from south-western China. These two strains had similar intra-strain SNP frequencies (1.104 and 1.278 % per nucleotide, respectively). The combined analysis revealed that 14 of the 20 examined fragments contained SNPs, ranging from two to 47 per fragment, and yielding a total of 178 SNPs out of the 10 428 sequenced nucleotides (an SNP frequency of 1.707 %). Among the 178 SNPs, one site had three alternative nucleotides, while the remaining 177 had two each, with 148 transitions and 29 transversions, resulting in a combined transition to transversion ratio of 5.1. In addition, the haplotype phases of all SNPs within individual fragments for both strains were determined. Phylogenetic analyses of these haplotypes revealed three kinds of haplotype relationship, including haplotype sharing both within and between strains. Furthermore, a subset of the SNPs detectable by restriction enzyme digests was screened for its distribution among 31 additional wild strains from five distinct locations in south-western China. The implications of these SNPs and haplotypes for our understanding of the genetics, population history, ecology and evolution of this important mushroom species are discussed.
-
-
-
-
Phylogeny of the alpha and beta subunits of the dissimilatory adenosine-5′-phosphosulfate (APS) reductase from sulfate-reducing prokaryotes – origin and evolution of the dissimilatory sulfate-reduction pathway
More LessNewly developed PCR assays were used to PCR-amplify and sequence fragments of the dissimilatory adenosine-5′-phosphosulfate (APS) reductase genes (aprBA) comprising nearly the entire gene locus (2·2–2·4 kb, equal to 92–94 % of the protein coding sequence) from 75 sulfate-reducing prokaryotes (SRP) of a taxonomically wide range. Comparative phylogenetic analysis included all determined and publicly available AprBA sequences from SRP and selected homologous sequences of sulfur-oxidizing bacteria (SOB). The almost identical AprB and AprA tree topologies indicated a shared evolutionary path for the aprBA among the investigated SRP by vertical inheritance and concomitant lateral gene transfer (LGT). The topological comparison of AprB/A- and 16S rRNA gene-based phylogenetic trees revealed novel LGT events across the SRP divisions. Compositional gene analysis confirmed Thermacetogenium phaeum to be the first validated strain affected by a recent lateral transfer of aprBA as a putative effect of long-term co-cultivation with a Thermodesulfovibrio species. Interestingly, the Apr proteins of SRP and SOB diverged into two phylogenetic lineages, with the SRP affiliated with the green sulfur bacteria, e.g. Chlorobaculum tepidum, while the Allochromatium vinosum-related sequences formed a distinct group. Analysis of genome data indicated that this phylogenetic separation is also reflected in the differing presence of the putative proteins functionally associated with Apr, QmoABC complex (quinone-interacting membrane-bound oxidoreductase) and AprM (transmembrane protein). Scenarios for the origin and evolution of the dissimilatory APS reductase are discussed within the context of the dissimilatory sulfite reductase (DsrAB) phylogeny, the appearance of QmoABC and AprM in the SRP and SOB genomes, and the geochemical setting of Archean Earth.
-
-
-
Bartonella henselae exists as a mosaic of different genetic variants in the infected host
More LessBartonella henselae is a fastidious bacterium associated with infections in humans and cats. The mechanisms involved in the long-term survival of bartonellae despite vigorous host immune responses are poorly understood. Generation of genetic variants is a possible strategy to circumvent the host specific immune responses. The authors have recently demonstrated the coexistence of different genetic variants within the progeny of three primary B. henselae isolates from Berlin by PFGE analysis. Aims of the present study were to determine whether coexistence of different variants is a common feature of B. henselae isolates worldwide and whether the genetic variants originally emerged in vivo. Thirty-four primary isolates from different geographical regions were analysed by subjecting multiple single-colony-derived cultures to PFGE analysis. Up to three genetic variants were detected within 20 (58.8 %) isolates, indicating that most primary isolates display a mosaic-like structure. The close relatedness of the genetic variants within an isolate was confirmed by multi-locus sequence typing. In contrast to the primary isolates, no genetic variants were detected within the progeny of 20 experimental clones generated in vitro from 20 primary isolates, suggesting that the variants were not induced in vitro during the procedure of PFGE analysis. Hence, the genetic variants within a primary isolate most likely originally emerged in vivo. Consideration of the mosaic structure of primary isolates is essential when interpreting typing studies on B. henselae.
-
-
-
Comparative genomic hybridization and physiological characterization of environmental isolates indicate that significant (eco-)physiological properties are highly conserved in the species Escherichia coli
More LessEscherichia coli, the common inhabitant of the mammalian intestine, exhibits considerable intraspecies genomic variation, which has been suggested to reflect adaptation to different ecological niches. Also, regulatory trade-offs, e.g. between catabolic versatility and stress protection, are thought to result in significant physiological differences between strains. For these reasons, the relevance of experimental observations made for ‘domesticated’ E. coli strains with regard to the behaviour of this species in its natural environments is often questioned and doubts are frequently raised on the status of E. coli as a defined species. The variability of important (eco-)physiological functions, such as carbon substrate uptake and breakdown capabilities, as well as stress defence mechanisms, in the genomes of commensal and pathogenic E. coli strains were therefore investigated. Furthermore, (eco-)physiological properties of environmental strains were compared to standard laboratory strain K-12 MG1655. Catabolic, stress protection, and carbon- and energy source transport operons showed a very low intraspecies variability in 57 commensal and pathogenic E. coli. Environmental isolates adapted to glucose-limited growth in a similar way as E. coli MG1655, namely by increasing their catabolic flexibility and by inducing high-affinity substrate uptake systems. The results obtained indicate that significant (eco-)physiological properties are highly conserved in the natural population of E. coli. This questions the proposed dominant role of horizontal gene transfer for niche adaptation.
-
-
-
Convergent evolution of phytopathogenic pseudomonads onto hazelnut
More LessPseudomonas syringae pv. avellanae (synonym: P. avellanae, Pav) is the causal agent of hazelnut decline in Greece and Italy. The population structure and evolutionary relationships of 22 strains from these two countries were examined by multilocus sequence typing (MLST) of four housekeeping genes (gapA, gltA, gyrB and rpoD). Neighbour-joining and maximum-likelihood phylogenetic analysis revealed that Greek strains isolated from the original 1976 outbreak of hazelnut decline through 1990 were very similar to Italian strains isolated from 2002 through 2004. Other Italian strains that were isolated during the 1990s were very homogeneous and clustered in a clade that was quite distinct from the Greek isolates and Italian isolates from the 2000s. A split decomposition analysis found evidence for recombination between these two highly divergent clades in two of the four MLST housekeeping genes. Incorporating these data into a broad MLST analysis of the P. syringae species complex showed that the Pav Greek and Italian strains from the 2000s clustered with P. syringae phylogroup 1, which is predominantly composed of pathogens of tomato and Brassicaceae hosts, while the Pav Italian strains from the 1990s clustered in P. syringae phylogroup 2 and are most closely related to pea (Pisum sativum L.) pathogens. These results clearly indicate that the ability to infect hazelnuts has arisen twice. This evolutionary process may be due to de novo adaptation to hazelnut by local P. syringae strains (such as the colonizers of Leguminosae crops), or the result of genetic exchange from the original Greek Pav clonal group into a phylogroup 2 strain. The latter explanation is intriguing since there is no exchange of hazelnut propagative material between Italy and Greece, which would be a likely vector for the movement of these pathogens.
-
- Environmental Microbiology
-
-
-
Growth of Vibrio cholerae O1 Ogawa Eltor in freshwater
More LessGrowth of Vibrio cholerae O1 Ogawa Eltor was studied with a growth assay in which autoclaved and filtered (0.22 μm) freshwater was inoculated at low cell density (5×103 cells ml−1) and proliferation was followed with flow cytometry. Against the common view, V. cholerae was able to grow extensively in different kinds of freshwater. The bacterium multiplied in river water, lake water and effluent of a wastewater treatment plant up to a cell density of 1.55×106 cells ml−1. In these samples, apparent assimilable organic carbon (AOCapp) concentrations ranged from 52 up to 800 μg l−1 and the results demonstrate a positive trend between the AOCapp concentration and final cell concentration, suggesting that AOC was a key parameter governing growth of V. cholerae. No growth was observed in waters (tap and bottled drinking water) containing less than approximately 60 μg AOCapp l−1. When pure cultures of V. cholerae were grown on identical lake water at different temperatures (20, 25 and 30 °C) the maximum specific growth rates (μmax) achieved were 0.22 h−1, 0.32 h−1 and 0.45 h−1, respectively. In addition, growth was characterized in lake water samples amended with different concentrations of NaCl. The highest μmax of V. cholerae was recorded at moderate salinity levels (5 g NaCl l−1, μmax=0.84 h−1), whereas at 30 g NaCl l−1 (μmax=0.30 h−1) or 0 g NaCl l−1 (μmax=0.40 h−1) specific growth rates were significantly reduced. In the water tested here, μmax of V. cholerae was always around 50 % of that exhibited by a freshwater community of indigenous bacteria enriched from the water sampling site. Direct batch competition experiments between V. cholerae and the lake water bacterial community were performed at different temperatures in which V. cholerae was enumerated in the total community using fluorescent-surface antibodies. In all cases V. cholerae was able to grow and constituted around 10 % of the final total cell concentration of the community. No significant effect of temperature was observed on the outcome of the competition. Mathematical modelling of the competition at the different temperatures based on the calculated μmax values confirmed these experimental observations. The results demonstrate that V. cholerae is not only able to survive, but also able to grow in freshwater samples. In these experiments the bacterium was able to use a large fraction (12–62 %) of the AOCapp available to the bacterial AOC-test community, indicating that V. cholerae has the ability to gain access to the substrates present in freshwater even in competition with an autochthonous bacterial lake water consortium.
-
-
-
-
A novel degradation pathway in the assimilation of phenanthrene by Staphylococcus sp. strain PN/Y via meta-cleavage of 2-hydroxy-1-naphthoic acid: formation of trans-2,3-dioxo-5-(2′-hydroxyphenyl)-pent-4-enoic acid
More LessStaphylococcus sp. strain PN/Y, capable of utilizing phenanthrene as a sole source of carbon and energy, was isolated from petroleum-contaminated soil. In the degradation of phenanthrene by strain PN/Y, various metabolites, isolated and identified by a combination of chromatographic and spectrometric analyses, revealed a novel phenanthrene assimilation pathway involving 2-hydroxy-1-naphthoic acid. Metabolism of phenanthrene was initiated by the dioxygenation on the 1,2-position of phenanthrene followed by meta-cleavage of phenanthrene-1,2-diol, leading to 2-hydroxy-1-naphthoic acid, which was then processed via a novel meta-cleavage pathway, leading to the formation of trans-2,3-dioxo-5-(2′-hydroxyphenyl)-pent-4-enoic acid and subsequently to salicylic acid. In the lower pathway, salicylic acid was transformed to catechol, which was then metabolized by catechol-2,3-dioxygenase to 2-hydroxymuconaldehyde acid, ultimately forming TCA cycle intermediates. The catabolic genes involved in phenanthrene degradation were found to be plasmid-encoded. This detailed study of polycyclic aromatic hydrocarbon (PAH) metabolism by a Gram-positive species involving a unique ring-cleavage dioxygenase in a novel phenanthrene degradation pathway provides a new insight into the microbial degradation of PAHs.
-
Volumes and issues
-
Volume 170 (2024)
-
Volume 169 (2023)
-
Volume 168 (2022)
-
Volume 167 (2021)
-
Volume 166 (2020)
-
Volume 165 (2019)
-
Volume 164 (2018)
-
Volume 163 (2017)
-
Volume 162 (2016)
-
Volume 161 (2015)
-
Volume 160 (2014)
-
Volume 159 (2013)
-
Volume 158 (2012)
-
Volume 157 (2011)
-
Volume 156 (2010)
-
Volume 155 (2009)
-
Volume 154 (2008)
-
Volume 153 (2007)
-
Volume 152 (2006)
-
Volume 151 (2005)
-
Volume 150 (2004)
-
Volume 149 (2003)
-
Volume 148 (2002)
-
Volume 147 (2001)
-
Volume 146 (2000)
-
Volume 145 (1999)
-
Volume 144 (1998)
-
Volume 143 (1997)
-
Volume 142 (1996)
-
Volume 141 (1995)
-
Volume 140 (1994)
-
Volume 139 (1993)
-
Volume 138 (1992)
-
Volume 137 (1991)
-
Volume 136 (1990)
-
Volume 135 (1989)
-
Volume 134 (1988)
-
Volume 133 (1987)
-
Volume 132 (1986)
-
Volume 131 (1985)
-
Volume 130 (1984)
-
Volume 129 (1983)
-
Volume 128 (1982)
-
Volume 127 (1981)
-
Volume 126 (1981)
-
Volume 125 (1981)
-
Volume 124 (1981)
-
Volume 123 (1981)
-
Volume 122 (1981)
-
Volume 121 (1980)
-
Volume 120 (1980)
-
Volume 119 (1980)
-
Volume 118 (1980)
-
Volume 117 (1980)
-
Volume 116 (1980)
-
Volume 115 (1979)
-
Volume 114 (1979)
-
Volume 113 (1979)
-
Volume 112 (1979)
-
Volume 111 (1979)
-
Volume 110 (1979)
-
Volume 109 (1978)
-
Volume 108 (1978)
-
Volume 107 (1978)
-
Volume 106 (1978)
-
Volume 105 (1978)
-
Volume 104 (1978)
-
Volume 103 (1977)
-
Volume 102 (1977)
-
Volume 101 (1977)
-
Volume 100 (1977)
-
Volume 99 (1977)
-
Volume 98 (1977)
-
Volume 97 (1976)
-
Volume 96 (1976)
-
Volume 95 (1976)
-
Volume 94 (1976)
-
Volume 93 (1976)
-
Volume 92 (1976)
-
Volume 91 (1975)
-
Volume 90 (1975)
-
Volume 89 (1975)
-
Volume 88 (1975)
-
Volume 87 (1975)
-
Volume 86 (1975)
-
Volume 85 (1974)
-
Volume 84 (1974)
-
Volume 83 (1974)
-
Volume 82 (1974)
-
Volume 81 (1974)
-
Volume 80 (1974)
-
Volume 79 (1973)
-
Volume 78 (1973)
-
Volume 77 (1973)
-
Volume 76 (1973)
-
Volume 75 (1973)
-
Volume 74 (1973)
-
Volume 73 (1972)
-
Volume 72 (1972)
-
Volume 71 (1972)
-
Volume 70 (1972)
-
Volume 69 (1971)
-
Volume 68 (1971)
-
Volume 67 (1971)
-
Volume 66 (1971)
-
Volume 65 (1971)
-
Volume 64 (1970)
-
Volume 63 (1970)
-
Volume 62 (1970)
-
Volume 61 (1970)
-
Volume 60 (1970)
-
Volume 59 (1969)
-
Volume 58 (1969)
-
Volume 57 (1969)
-
Volume 56 (1969)
-
Volume 55 (1969)
-
Volume 54 (1968)
-
Volume 53 (1968)
-
Volume 52 (1968)
-
Volume 51 (1968)
-
Volume 50 (1968)
-
Volume 49 (1967)
-
Volume 48 (1967)
-
Volume 47 (1967)
-
Volume 46 (1967)
-
Volume 45 (1966)
-
Volume 44 (1966)
-
Volume 43 (1966)
-
Volume 42 (1966)
-
Volume 41 (1965)
-
Volume 40 (1965)
-
Volume 39 (1965)
-
Volume 38 (1965)
-
Volume 37 (1964)
-
Volume 36 (1964)
-
Volume 35 (1964)
-
Volume 34 (1964)
-
Volume 33 (1963)
-
Volume 32 (1963)
-
Volume 31 (1963)
-
Volume 30 (1963)
-
Volume 29 (1962)
-
Volume 28 (1962)
-
Volume 27 (1962)
-
Volume 26 (1961)
-
Volume 25 (1961)
-
Volume 24 (1961)
-
Volume 23 (1960)
-
Volume 22 (1960)
-
Volume 21 (1959)
-
Volume 20 (1959)
-
Volume 19 (1958)
-
Volume 18 (1958)
-
Volume 17 (1957)
-
Volume 16 (1957)
-
Volume 15 (1956)
-
Volume 14 (1956)
-
Volume 13 (1955)
-
Volume 12 (1955)
-
Volume 11 (1954)
-
Volume 10 (1954)
-
Volume 9 (1953)
-
Volume 8 (1953)
-
Volume 7 (1952)
-
Volume 6 (1952)
-
Volume 5 (1951)
-
Volume 4 (1950)
-
Volume 3 (1949)
-
Volume 2 (1948)
-
Volume 1 (1947)
Most Read This Month
