 -
Volume 149,
Issue 10,
2003
-
Volume 149,
Issue 10,
2003

Volume 149, Issue 10, 2003
- Life And Times Of A Superbug: Staphylococcus Research
-
-
-
The adhesive and immunomodulating properties of the multifunctional Staphylococcus aureus protein Eap
More LessAdherence of Staphylococcus aureus to the host tissue is an important step in the initiation of pathogenesis. At least 10 adhesins produced by S. aureus have been described and it is becoming clear that the expression of these adhesins and their interactions with eukaryotic cells involve complex processes. Some of these, such as the fibronectin-binding proteins (FnBPs) and Clumping Factor A, are well characterized. However, in the last 10 years a number of novel S. aureus adhesins have been described. Functional analyses of these proteins, one of which is Eap (extracellular adherence protein, also known as Map and p70), are revealing important information on the pathogenesis of staphylococcal disease. More than 10 years after the first report of Eap, we are beginning to understand that this protein, which has a broad spectrum of functions, may be a critical factor in the pathogenesis of S. aureus. This review will focus on the interactions of Eap with eukaryotic cells, plasma proteins and the extracellular matrix as well as on the recently recognized role of Eap as an important mediator in the immune response to staphylococcal infection.
-
-
-
The staphylococcal saeRS system coordinates environmental signals with agr quorum sensing
More Lesssae is a two-component signal transduction system in Staphylococcus aureus that regulates the expression of many virulence factors at the transcriptional level and appears to act synergistically with agr in some cases. In this study, the interactions between sae and agr have been characterized in some detail. It was found that the sae locus is larger and more complex than originally envisioned, in that it is expressed from several promoters, giving rise to four or five transcripts, at least three of which are initiated upstream of saeRS and contain two additional reading frames, here designated saeP and saeQ, which are likely to have important roles in sae function. The upstream transcripts are induced during exponential phase concomitantly with the onset of RNAIII synthesis and their induction requires the agr effector, RNAIII, but is blocked by several environmental signals that override the effects of RNAIII. saeR is also required for the induction of these transcripts, so that the sae locus contains an autoinduction circuit. It is suggested that sae is downstream of agr in the exoprotein activation pathway (and also epistatic with agr), that it coordinates the effects of environmental signals with the agr quorum-sensing system, and therefore that it is a key intermediary in the overall regulatory strategy by which S. aureus senses and responds to its environment.
-
-
-
Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon
More LessThe molecular events following inhibition of bacterial peptidoglycan synthesis have not been studied extensively. Previous proteomic studies have revealed that certain proteins are produced in increased amounts upon challenge of Staphylococcus aureus with cell-wall-active antibiotics. In an effort to further those studies, the genes upregulated in their expression in response to cell-wall-active antibiotics have been identified by genome-wide transcriptional profiling using custom-made Affymetrix S. aureus GeneChipsTM. A large number of genes, including ones encoding proteins involved in cell-wall metabolism (including pbpB, murZ, fmt and vraS) and stress responses (including msrA, htrA, psrA and hslO), were upregulated by oxacillin, d-cycloserine or bacitracin. This response may represent the transcriptional signature of a cell-wall stimulon induced in response to cell-wall-active agents. The findings imply that treatment with cell-wall-active antibiotics results in damage to proteins including oxidative damage. Additional genes in a variety of functional categories were upregulated uniquely by each of the three cell-wall-active antibiotics studied. These changes in gene expression can be viewed as an attempt by the organism to defend itself against the antibacterial activities of the agents.
-
-
-
Application of a bacterial two-hybrid system for the analysis of protein–protein interactions between FemABX family proteins
More LessProtein–protein interactions play an important role in all cellular processes. The development of two-hybrid systems in yeast and bacteria allows for in vivo assessment of such interactions. Using a recently developed bacterial two-hybrid system, the interactions of the Staphylococcus aureus proteins FemA, FemB and FmhB, members of the FemABX protein family, which is involved in peptidoglycan biosynthesis and β-lactam resistance of numerous Gram-positive bacteria, were analysed. While FmhB is involved in the addition of glycine 1 of the pentaglycine interpeptide of S. aureus peptidoglycan, FemA and FemB are specific for glycines 2/3 and 4/5, respectively. FemA–FemA, FemA–FemB and FemB–FemB interactions were found, while FmhB exists solely as a monomer. Interactions detected by the bacterial two-hybrid system were confirmed using the glutathione S-transferase-pulldown assay and gel filtration.
-
-
-
Multiple methionine sulfoxide reductase genes in Staphylococcus aureus: expression of activity and roles in tolerance of oxidative stress
More LessStaphylococcus aureus contains three genes encoding MsrA-specific methionine sulfoxide reductase (Msr) activity (msrA1, msrA2 and msrA3) and an additional gene that encodes MsrB-specific Msr activity. Data presented here suggest that MsrA1 is the major contributor of the MsrA activity in S. aureus. In mutational analysis, while the total Msr activity in msrA2 mutant was comparable to that of the parent, Msr activity was significantly up-regulated in the msrA1 or msrA1 msrA2 double mutant. Assessment of substrate specificity together with increased reactivity of the cell-free protein extracts of the msrA1 mutants to anti-MsrB polyclonal antibodies in Western analysis provided evidence that increased Msr activity was due to elevated synthesis of MsrB in the MsrA1 mutants. Previously, it was reported that oxacillin treatment of S. aureus cells led to induced synthesis of MsrA1 and a mutation in msrA1 increased the susceptibility of the organism to H2O2. A mutation in the msrA2 gene, however, was not significant for the bacterial oxidative stress response. In complementation assays, while the msrA2 gene was unable to complement the msrA1 msrA2 double mutant for H2O2 resistance, the same gene restored H2O2 tolerance in the double mutant when placed under the control of the msrA1 promoter. However, msrA1 which was able to complement the oxidative stress response in msrA1 mutants could not restore the tolerance of the msrA1 msrA2 mutants to H2O2 when placed under the control of the msrA2 promoter. Additionally, although the oxacillin minimum inhibitory concentration of the msrA1 mutant was comparable to that of the wild-type parent, in shaking liquid culture, the msrA1 mutant responded more efficiently to sublethal doses of oxacillin. The data suggest complex regulation of Msr proteins and a more significant physiological role for msrA1/msrB in S. aureus.
-
-
-
Role and regulation of the superoxide dismutases of Staphylococcus aureus
More LessStaphylococcus aureus has two superoxide dismutases (SODs), encoded by the sodA and sodM genes, which inactivate harmful superoxide radicals (
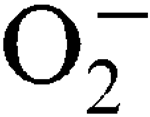 ) encountered during host infection or generated from aerobic metabolism. The transcriptional start sites have been mapped and expression analysis on reporter fusions in both genes has been carried out. Under standard growth conditions, manganese (Mn), a mineral superoxide scavenger, elevated total SOD activity but had no effect on the transcription of either gene. Transcription of sodA and sodM was most strongly induced by either internally or externally generated
) encountered during host infection or generated from aerobic metabolism. The transcriptional start sites have been mapped and expression analysis on reporter fusions in both genes has been carried out. Under standard growth conditions, manganese (Mn), a mineral superoxide scavenger, elevated total SOD activity but had no effect on the transcription of either gene. Transcription of sodA and sodM was most strongly induced by either internally or externally generated 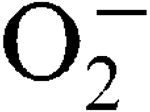 , respectively. Sensitivity to internally generated
, respectively. Sensitivity to internally generated 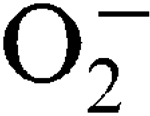 was linked with SodA deficiency. Mn supplementation completely rescued a sodA mutant when challenged by internally generated
was linked with SodA deficiency. Mn supplementation completely rescued a sodA mutant when challenged by internally generated 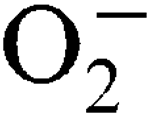 , and this was growth-phase-dependent. Sensitivity to externally generated
, and this was growth-phase-dependent. Sensitivity to externally generated 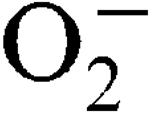 stress was only observed in a sodA sodM mutant and was Mn-independent. In a mouse abscess model of infection, isogenic sodA, sodM and sodA
sodM mutants had reduced virulence compared to the parental strain, showing the importance of the enzymic
stress was only observed in a sodA sodM mutant and was Mn-independent. In a mouse abscess model of infection, isogenic sodA, sodM and sodA
sodM mutants had reduced virulence compared to the parental strain, showing the importance of the enzymic 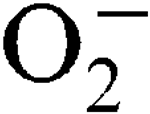 scavenging system for the survival of the pathogen.
scavenging system for the survival of the pathogen.
-
-
-
The Staphylococcus aureus surface protein SasG and its homologues promote bacterial adherence to human desquamated nasal epithelial cells
More LessStaphylococcus aureus binds to human desquamated nasal epithelial cells, a phenomenon likely to be important in nasal colonization. ClfB was identified previously as one staphylococcal adhesin that promoted binding to nasal epithelia. In this study, it is shown that the S. aureus surface protein SasG, identified previously by in silico analysis of genome sequences, and two homologous proteins, Pls of S. aureus and AAP of Staphylococcus epidermidis, also promote bacterial adherence to nasal epithelial cells. Conditions for in vitro expression of SasG by S. aureus were not found. Adherence assays were therefore performed with S. aureus and Lactococcus lactis expressing SasG from an expression plasmid. These studies showed that SasG did not bind several ligands typically bound by S. aureus. Significantly, SasG and Pls did promote bacterial adherence to nasal epithelial cells. Furthermore, pre-incubation of epithelial cells with purified recombinant proteins revealed that the N-terminal A regions of SasG, Pls and AAP, but not the B repeats of SasG, inhibited adherence of L. lactis expressing SasG in a dose-dependent fashion. These results suggest that SasG, Pls and AAP bind to the same as-yet-unidentified receptor on the surface of nasal epithelial cells. In addition, expression of SasG, like Pls, reduced adherence of S. aureus to fibronectin and fibrinogen.
-
-
-
Identification and characterization of a novel autolysin (Aae) with adhesive properties from Staphylococcus epidermidis
More LessStaphylococcus epidermidis biofilm formation on polymer surfaces is considered a major pathogenicity factor in foreign-body-associated infections. Previously, the 148 kDa autolysin AtlE from S. epidermidis, which is involved in the initial attachment of the cells to polymer surfaces and also binds to the extracellular matrix protein vitronectin, was characterized. Here, the characterization of a novel autolysin/adhesin (Aae) in S. epidermidis is described. Aae was identified as a 35 kDa surface-associated protein that has bacteriolytic activity and binds vitronectin. Its N-terminal amino acid sequence was determined and the respective gene, aae, was cloned. DNA-sequence analysis revealed that aae encodes a deduced protein of 324 amino acids with a predicted molecular mass of 35 kDa. Aae contains three repetitive sequences in its N-terminal portion. These repeats comprise features of a putative peptidoglycan binding domain (LysM domain) found in a number of enzymes involved in cell-wall metabolism and also in some adhesins. Expression of aae by Escherichia coli and subsequent analysis revealed that Aae possesses bacteriolytic activity and adhesive properties. The interaction of Aae with fibrinogen, fibronectin and vitronectin was found to be dose-dependent and saturable and to occur with high affinity, by using the real-time Biomolecular Interaction Analysis (BIA). Aae binds to the Aα- and Bβ-chains of fibrinogen and to the 29 kDa N-terminal fragment of fibronectin. In conclusion, Aae is a surface-associated protein with bacteriolytic and adhesive properties representing a new member of the staphylococcal autolysin/adhesins potentially involved in colonization.
-
- Cell And Developmental Biology
-
-
-
Aggregation of yeast cells: direct measurement of discrete lectin–carbohydrate interactions
More LessAggregation of microbial cells mediated by specific interactions plays a pivotal role in the natural environment, in medicine and in biotechnological processes. Here we used atomic force microscopy (AFM) to measure individual lectin–carbohydrate interactions involved in the flocculation of yeast cells, an aggregation event of crucial importance in fermentation technology. AFM probes functionalized with oligoglucose carbohydrates were used to record force-distance curves on living yeast cells at a rate of 0·5 μm s−1. Flocculating cells showed adhesion forces of 121±53 pN, reflecting the specific interaction between individual cell-surface lectins and glucose residues. Similar adhesion forces, 117±41 pN, were measured using probes functionalized with the lectin concanavalin A and attributed to specific binding to cell-surface mannose residues. By contrast, specific interaction forces were not observed in non-flocculating conditions, i.e. in the presence of mannose or when using non-flocculating cells, pointing to their involvement in yeast flocculation. The single molecule force spectroscopy measurements presented here provide a means to study a variety of cellular interactions at the molecular level, such as the adhesion of bacteria to animal and plant tissues.
-
-
-
-
The cAMP phosphodiesterase encoded by CaPDE2 is required for hyphal development in Candida albicans
More LessThe cAMP-dependent pathway, which regulates yeast-to-hypha morphogenesis in Candida albicans, is controlled by changes in cAMP levels determined by the processes of synthesis and hydrolysis. Both low- and high-affinity cAMP phosphodiesterases are encoded in the C. albicans genome. CaPDE2, encoding the high-affinity cAMP phosphodiesterase, has been cloned and shown to be toxic in Saccharomyces cerevisiae upon overexpression under pGAL1, but functional under the moderate pMET3. Deletion of CaPDE2 causes elevated cAMP levels and responsiveness to exogenous cAMP, higher sensitivity to heat shock, severe growth defects at 42 °C and highly reduced levels of EFG1 transcription. In vitro in hypha-inducing liquid medium CaPDE2, deletion prohibits normal hyphal, but not pseudohyphal growth. On solid medium capde2 mutants form aberrant hyphae, with fewer branches and almost no lateral buds, which are deficient in hypha-to-yeast reversion. The phenotypic defects of capde2 mutants show that the cAMP-dependent pathway plays specific roles in hyphal and pseudohyphal development, its regulatory role however, being greater in liquid than on solid medium in vitro. The increased expression of CaPDE2 after serum addition correlates well with a drop in cAMP levels following the initial rise in response to the hyphal inducer. These results suggest that Capde2p mediates a desensitization mechanism by lowering basal cAMP levels in response to environmental stimuli in C. albicans.
-
-
-
Bacillus subtilis spoVIF (yjcC) gene, involved in coat assembly and spore resistance
More LessIn systematic screening four sporulation-specific genes, yjcA, yjcB, yjcZ and yjcC, of unknown function were found in Bacillus subtilis. These genes are located just upstream of the cotVWXYZ gene cluster oriented in the opposite direction. Northern blot analysis showed that yjcA was transcribed by the SigE RNA polymerase beginning 2 h (t 2) after the onset of sporulation, and yjcB, yjcZ and yjcC were transcribed by the SigK RNA polymerase beginning at t 4 of sporulation. The transcription of yjcZ was dependent on SigK and GerE. The consensus sequences of the appropriate sigma factors were found upstream of each gene. There were putative GerE-binding sites upstream of yjcZ. Insertional inactivation of the yjcC gene resulted in a reduction in resistance of the mutant spores to lysozyme and heat. Transmission electron microscopic examination of yjcC spores revealed a defect of sporulation at stage VI, resulting in loss of spore coats. These results suggest that YjcC is involved in assembly of spore coat proteins that have roles in lysozyme resistance. It is proposed that yjcC should be renamed as spoVIF.
-
- Biochemistry And Molecular Biology
-
-
-
Proposed pathway for the biosynthesis of serovar-specific glycopeptidolipids in Mycobacterium avium serovar 2
More LessMembers of the Mycobacterium avium complex are distinguished by the presence of highly antigenic surface molecules called glycopeptidolipids (GPLs) and the oligosaccharide portion of the serovar-specific GPL defines the 28 serovars. Previously, the genomic region (ser2) encoding the enzymes responsible for the glycosylation of the lipopeptide core to generate the serovar-2-specific GPLs has been described. In this work, the ser2 gene clusters of M. avium serovar 2 strains 2151 and TMC 724 were fully sequenced and compared to the homologous regions of M. avium serovar 1 strain 104, M. avium subsp. paratuberculosis and M. avium subsp. silvaticum. It was also determined that 104Rg, a mutant of strain 104 that produces truncated GPLs, lost several GPL biosynthesis genes by deletion. This comparison, together with analysis of protein similarities, supports a biosynthetic model in which serovar-2-specific GPLs are synthesized from a serovar-1-specific GPL intermediate that is derived from a non-specific GPL precursor. We also identified a gene encoding an enzyme that is necessary for the biosynthesis of serovar-3- and 9-specific GPLs, but not serovar-2-specific GPLs, suggesting that the different serovars may have evolved from the acquisition or loss of genetic information. In addition, a subcluster of genes for the biosynthesis and transfer of fucose, which are needed to make serovar-specific GPLs such as those of serovar 2, is found in the non-GPL-producing M. avium subspecies paratuberculosis and silvaticum.
-
-
-
-
Activation of hilA expression at low pH requires the signal sensor CpxA, but not the cognate response regulator CpxR, in Salmonella enterica serovar Typhimurium
More LessA two-component regulatory system, cpxR–cpxA, plays an important role in the pH-dependent regulation of virF, a global activator for virulence determinants including invasion genes, in Shigella sonnei. The authors examined whether the cpxR–cpxA homologues have some function in the expression of Salmonella enterica serovar Typhimurium invasion genes via the regulation of hilA, an activator for these genes. In a Salmonella cpxA mutant, the hilA expression level was reduced to less than 10 % of that in the parent strain at pH 6·0. This mutant strain also showed undetectable synthesis of an invasion gene product, SipC, at pH 6·0 and reduced cell invasion capacity – as low as 20 % of that of the parent. In this mutant, the reduction in hilA expression was much less marked at pH 8·0 than at pH 6·0 – no less than 50 % of that in the parent, and no significant reduction was observed in either SipC synthesis or cell invasion rate, compared to the parent. Unexpectedly, a Salmonella cpxR mutant strain and the parent showed no apparent difference in all three characteristics described above at either pH. These results indicate that in Salmonella, the sensor kinase CpxA activates hilA, and consequently, invasion genes and cell invasion capacity at pH 6·0. At pH 8·0, however, CpxA does not seem to have a large role in activation of these factors. Further, the results show that this CpxA-mediated activation does not require its putative cognate response regulator, CpxR. This suggests that CpxA may interact with regulator(s) other than CpxR to achieve activation at low pH.
-
-
-
The curli biosynthesis regulator CsgD co-ordinates the expression of both positive and negative determinants for biofilm formation in Escherichia coli
More LessProduction of curli, extracellular structures important for biofilm formation, is positively regulated by OmpR, which constitutes with the EnvZ protein an osmolarity-sensing two-component regulatory system. The expression of curli is cryptic in most Escherichia coli laboratory strains such as MG1655, due to the lack of csgD expression. The csgD gene encodes a transcription activator of the curli-subunit-encoding csgBA operon. The ompR234 up-mutation can restore csgD expression, resulting in curli production and increased biofilm formation. In this report, it is shown that ompR234-dependent csgD expression, in addition to csgBA activation during stationary phase of growth, stimulates expression of the yaiC gene and negatively regulates at least two other genes, pepD and yagS. The promoter regions of these four genes share a conserved 11 bp sequence (CGGGKGAKNKA), necessary for csgBA and yaiC regulation by CsgD. While at both the csgBA and yaiC promoters the sequence is located upstream of the promoter elements, in both yagS and pepD it overlaps either the putative −10 sequence or the transcription start point, suggesting that CsgD can function as both an activator and a repressor. Adhesion experiments show that csgD-independent expression of both yagS and pepD from a multicopy plasmid negatively affects biofilm formation, which, in contrast, is stimulated by yaiC expression. Thus it is proposed that CsgD stimulates biofilm formation in E. coli by contemporary activation of adhesion positive determinants (the curli-encoding csg operons and the product of the yaiC gene) and repression of negative effectors such as yagS and pepD.
-
-
-
Identification of Pseudomonas proteins coordinately induced by acidic amino acids and their amides: a two-dimensional electrophoresis study
More LessThe acidic amino acids (Asp, Glu) and their amides (Asn, Gln) are excellent growth substrates for many pseudomonads. This paper presents proteomics data indicating that growth of Pseudomonas fluorescens ATCC 13525 and Pseudomonas putida KT2440 on these amino acids as sole source of carbon and nitrogen leads to the induction of a defined set of proteins. Using mass spectrometry and N-terminal sequencing, a number of these proteins were identified as enzymes and transporters involved in amino acid uptake and metabolism. Most of them depended on the alternative sigma factor σ 54 for expression and were subject to strong carbon catabolite repression by glucose and citrate cycle intermediates. For a subset of the identified proteins, the observed regulatory effects were independently confirmed by RT-PCR. The authors propose that the respective genes (together with others still to be identified) make up a regulon that mediates uptake and utilization of the abovementioned amino acids.
-
-
-
Disruption of the gene encoding the ChiB1 chitinase of Aspergillus fumigatus and characterization of a recombinant gene product
More LessThe gene encoding a major, inducible 45 kDa chitinase of Aspergillus fumigatus was cloned and analysis of the deduced amino acid sequence identified a chitinase of the fungal/bacterial class which was designated ChiB1. Recombinant ChiB1, expressed in Pichia pastoris, was shown to function by a retaining mechanism of action. That is, the β-conformation of the chitin substrate linkage was preserved in the product in a manner typical of family 18 chitinases. Cleavage patterns with the N-acetylglucosamine (GlcNAc) oligosaccharide substrates GlcNAc4, GlcNAc5 and GlcNAc6 indicated that the predominant reaction involved hydrolysis of GlcNAc2 from the non-reducing end of each substrate. Products of transglycosylation were also identified in each incubation. Following disruption of chiB1 by gene replacement, growth and morphology of disruptants and of the wild-type strain were essentially identical. However, during the autolytic phase of batch cultures the level of chitinase activity in culture filtrate from a disruptant was much lower than the activity from the wild-type. The search for chitinases with morphogenetic roles in filamentous fungi should perhaps focus on chitinases of the fungal/plant class although such an investigation will be complicated by the identification of at least 11 putative active site domains for family 18 chitinases in the A. fumigatus TIGR database (http://www.tigr.org/).
-
-
-
A novel tannase from Aspergillus niger with β-glucosidase activity
More LessAn extracellular tannase was produced from solid-state cultures of Aspergillus niger. The enzyme was purified to homogeneity from the cell-free culture broth by preparative isoelectric focusing and by FPLC using anion-exchange and gel-filtration chromatography. SDS-PAGE analysis as well as gel localization studies of purified tannase indicated the presence of two enzyme forms, with molecular masses of 90 kDa and 180 kDa. The tannase had an isoelectric point of 3·8, a temperature optimum of 60–70 °C and a pH optimum of 6·0. The substrate specificity of the tannase was determined by HPLC analysis of tannin substrates and products. The enzyme was able to remove gallic acid from both condensed and hydrolysable tannins. Internal sequences were obtained from each of the gel-purified and trypsin-digested tannase forms. The peptide sequences obtained from both forms were identical to sequences within a β-glucosidase from Aspergillus kawachii. The purified tannase was tested for β-glucosidase activity and was shown to hydrolyse cellobiose efficiently. However, no β-glucosidase activity was detected when the enzyme was assayed in the presence of tannic acid.
-
-
-
Integrative, multifunctional plasmids for hypha-specific or constitutive expression of green fluorescent protein in Candida albicans
More LessThe authors have engineered plasmid constructs for developmental and constitutive expression of yeast-enhanced green fluorescent protein (yEGFP3) in Candida albicans. The promoter for the hyphae-specific gene Hyphal Wall Protein 1 (HWP1) conferred developmental expression of yEGFP3 in germ tubes and hyphae but not in yeasts or pseudohyphae when targeted to the ENO1 (enolase) locus in single copy. The pHWP1GFP3 construct allows for the easy visualization of HWP1 promoter activity in individual cells expressing true hyphae without having to prepare RNA for analysis. Constitutive expression of yEGFP was seen in all cell morphologies when the HWP1 promoter was replaced with the ENO1 promoter region. The use of the plasmids for expression of genes other than yEGFP3 was examined by substituting the putative C. albicans BCY1 (SRA1) gene, a component of the cAMP signalling pathway involved in yeast to hyphae transitions, for yEGFP3. Strains overexpressing BCY1 from the ENO1 promoter were inhibited in germ tube formation and filamentation in both liquid and solid media, a phenotype consistent with keeping protein kinase A in its inactive form by association with Bcy1p. The plasmids are suitable for studies of germ tube induction or assessing germ tube formation by measuring yEGFP3 expression, for inducible expression of genes concomitant with germ tube formation by the HWP1 promoter, for constitutive expression of genes by the ENO1 promoter, and for expressing yEGFP3 using a promoter of choice.
-
- Biodiversity And Evolution
-
-
-
Clonal population structure of Pseudomonas avellanae strains of different origin based on multilocus enzyme electrophoresis
More LessTo assess the genetic diversity and genetic relationships of Pseudomonas avellanae, the causative agent of hazelnut decline, a total of 102 strains, obtained from central Italy (provinces of Viterbo and Rome) and northern Greece, were studied using multilocus enzyme electrophoresis (MLEE). Their allelic variation in 10 loci was determined. All loci were polymorphic and 53 electrophoretic types (ETs) were identified from the total sample. The mean genetic diversity (H) was 0·65 and this value ranged from 0·37 for the least polymorphic to 0·82 for the most polymorphic locus. The dendrogram originated from MLEE data indicated two main groups of ETs, A and B. The groups do not appear to be correlated to the geographic origin of the strains, although all the ETs from northern Greece clustered into subgroup B1. Pseudomonas syringae pv. actinidiae and P. syringae pv. theae, included in the analysis as outgroups, clustered apart. The index of association (I A) for P. avellanae was 0·90. The I A values were always significantly different from zero for the population subsets studied and no epidemic structure was found. These results would indicate that the population structure of P. avellanae is clonal either in northern Greece or in central Italy. The recent outbreaks of the bacterium in new areas of hazelnut cultivation would explain the current clonal structure that is persisting over decades.
-
-
-
-
Allelic variation in the contiguous loci encoding Candida albicans ALS5, ALS1 and ALS9
More LessThe ALS gene family of Candida albicans consists of eight genes (ALS1 to ALS7 and ALS9) that encode cell-wall glycoproteins involved in adhesion to host surfaces. Considerable allelic sequence variability has been documented for regions of ALS genes encoding repeated sequences. Although regions of ALS genes encoding non-repeated sequences tend to be more conserved, some sequence divergence has been noted, particularly for alleles of ALS5. Data from the C. albicans genome sequencing project provided the first indication that strain SC5314 encoded two divergent ALS9-like sequences and that three of the ALS genes (ALS5, ALS1 and ALS9) were contiguous on chromosome 6. Data from PCR analysis and construction of both single and double deletion mutants indicated that the divergent sequences were alleles of ALS9, and located downstream of ALS5 and ALS1. Sequences within the 5′ domain of ALS9-1 and ALS9-2 varied by 11 %. Within the 3′ domain of each allele, extra nucleotides were present in two regions of ALS9-2, designated Variable Block 1 (VB1) and Variable Block 2 (VB2). Analysis of strains from the five major C. albicans genetic clades showed that both ALS9 alleles are widespread among these strains, that the sequences of ALS9-1 and ALS9-2 are conserved among diverse strains and that recombinant ALS9 alleles have been generated during C. albicans evolution. Phylogenetic analysis showed that, although divergent in sequence, ALS9 alleles are more similar to each other than to any other ALS genes. The degree of sequence divergence for ALS9 greatly exceeds that observed previously for other ALS genes and may result in functional differences for the proteins encoded by the two alleles.
-
- Environmental Microbiology
-
-
-
Degradation of alkanes and highly chlorinated benzenes, and production of biosurfactants, by a psychrophilic Rhodococcus sp. and genetic characterization of its chlorobenzene dioxygenase
More LessRhodococcus sp. strain MS11 was isolated from a mixed culture. It displays a diverse range of metabolic capabilities. During growth on 1,2,4-trichlorobenzene, 1,2,4,5-tetrachlorobenzene (1,2,4,5-TeCB) and 3-chlorobenzoate stoichiometric amounts of chloride were released. It also utilized all three isomeric dichlorobenzenes and 1,2,3-trichlorobenzene as the sole carbon and energy source. Furthermore, the bacterium grew well on a great number of n-alkanes ranging from n-heptane to n-triacontane and on the branched alkane 2,6,10,14-tetramethylpentadecane (pristane) and slowly on n-hexane and n-pentatriacontane. It was able to grow at temperatures from 5 to 30 °C, with optimal growth at 20 °C, and could tolerate 6 % NaCl in mineral salts medium. Genes encoding the initial chlorobenzene dioxygenase were detected by using a primer pair that was designed against the α-subunit (TecA1) of the chlorobenzene dioxygenase of Ralstonia (formerly Burkholderia) sp. strain PS12. The amino acid sequence of the amplified part of the α-subunit of the chlorobenzene dioxygenase of Rhodococcus sp. strain MS11 showed >99 % identity to the α-subunit of the chlorobenzene dioxygenase from Ralstonia sp. strain PS12 and the parts of both α-subunits responsible for substrate specificity were identical. The subsequent enzymes dihydrodiol dehydrogenase and chlorocatechol 1,2-dioxygenase were induced in cells grown on 1,2,4,5-TeCB. During cultivation on medium-chain-length n-alkanes ranging from n-decane to n-heptadecane, including 1-hexadecene, and on the branched alkane pristane, strain MS11 produced biosurfactants lowering the surface tension of the cultures from 72 to ⩽29 mN m−1. Glycolipids were extracted from the supernatant of a culture grown on n-hexadecane and characterized by 1H- and 13C-NMR-spectroscopy and mass spectrometry. The two major components consisted of α,α-trehalose esterified at C-2 or C-4 with a succinic acid and at C-2′ with a decanoic acid. They differed from one another in that one 2,3,4,2′-trehalosetetraester, found in higher concentration, was esterified at C-2, C-3 or C-4 with one octanoic and one decanoic acid and the other one, of lower concentration, with two octanoic acids. The results demonstrate that Rhodococcus sp. strain MS11 may be well suited for bioremediation of soils and sediments contaminated for a long time with di-, tri- and tetrachlorobenzenes as well as alkanes.
-
-
- Genes And Genomes
-
-
-
Transcriptional regulation of drug efflux genes by EvgAS, a two-component system in Escherichia coli
More LessA constitutively active mutant of histidine kinase sensor EvgS was found to confer multi-drug resistance (MDR) to an acrA-deficient Escherichia coli, indicating the relationship between the two-component system EvgAS and the expression of the MDR system. The observed MDR also depended on an outer-membrane channel, TolC. Microarray and S1 mapping assays indicated that, in the presence of this constitutive mutant EvgS, the level of transcription increased for some MDR genes, including the drug efflux genes emrKY, yhiUV, acrAB, mdfA and tolC. Transcription in vitro of emrK increased by the addition of phosphorylated EvgA. Transcription activation of tolC by the activated EvgS was, however, dependent on both EvgAS and PhoPQ (Mg2+-responsive two-component system), in agreement with the presence of the binding site (PhoP box) for the regulator PhoP in the tolC promoter region. Transcription in vitro of yhiUV also appears to require an as-yet-unidentified additional transcriptional factor besides EvgA. Taken together we propose that the expression of the MDR system is under a complex regulatory network, including the phosphorylated EvgA serving as the master regulator.
-
-
-
-
A cryptic plasmid of Yersinia enterocolitica encodes a conjugative transfer system related to the regions of CloDF13 Mob and IncX Pil
More LessYersinia enterocolitica 29930 (biotype 1A; O : 7,8), the producing strain of the phage-tail-like bacteriocin enterocoliticin, possesses a plasmid-encoded conjugative type IV transfer system. The genes of the conjugative system were found by screening of a cosmid library constructed from total DNA of strain 29930. The cosmid Cos100 consists of the vector SuperCos1 and an insert DNA of 40 303 bp derived from a cryptic plasmid of strain 29930. The conjugative transfer system consists of genes encoding a DNA transfer and replication system (Dtr) with close relationship to the mob region of the mobilizable plasmid CloDF13 and a gene cluster encoding a mating pair formation system (Mpf) closely related to the Mpf system of the IncX plasmid R6K. However, a gene encoding a homologue of TaxB, the coupling protein of the IncX system, is missing. The whole transfer region has a size of approximately 17 kb. The recombinant plasmid Cos100 was shown to be transferable between Escherichia coli and Yersinia with transfer frequencies up to 0·1 transconjugants per donor. Mutations generated by inserting a tetracycline cassette into putative tri genes yielded a transfer-deficient phenotype. Conjugative transfer of the cryptic plasmid could not be demonstrated in the original host Y. enterocolitica 29930. However, a kanamycin-resistance-conferring derivative of the plasmid was successfully introduced into E. coli K-12 by transformation and was shown to be self-transmissible. Furthermore, Southern blot hybridization and PCR experiments were carried out to elucidate the distribution of the conjugative transfer system in Yersinia. In total, six Y. enterocolitica biotype 1A strains harbouring closely related systems on endogenous plasmids were identified.
-
-
-
Transcript map of the temperate Lactobacillus gasseri bacteriophage ϕadh
More LessTemporal transcription of phage ϕadh was analysed during lytic reproduction. Based on Northern hybridizations the phage genome was divided into regions of early, middle and late transcription. Eight groups of overlapping transcripts, probably originating from common precursors, were distinguished. Early transcription of a 10·9 kb region adjacent to the lytic/lysogenic switch started within the first 10 min of infection and produced three groups of mRNAs mostly related to DNA replication. Four middle transcripts were observed 30 min after infection, corresponding to an 8·5 kb genomic region, which started at the replication origin (ori) and encompassed a DNA packaging function and the cos site. Three groups of late transcripts were first observed 50 min after infection, corresponding to a 21·1 kb region between the middle region and the attachment site (attP), encoding functions for capsid morphogenesis and host cell lysis. A fourth group of late-appearing mRNAs was divergently transcribed from the 3·2 kb section between attP and the lytic/lysogenic switch, including the repressor and integrase genes. Except for one set of early mRNAs, all the transcripts persisted until the end of the reproduction cycle. Two confirmed and two predicted promoters were assigned to transcript 5′ ends in the early region.
-
-
-
Identification of sporulation genes by genome-wide analysis of the σ E regulon of Bacillus subtilis
More LessDifferentiation in the spore-forming bacterium Bacillus subtilis is governed by the sequential activation of five sporulation-specific transcription factors. The early mother-cell-specific transcription factor, σ E, directs the transcription of many genes that contribute to the formation of mature, dormant spores. In this study, DNA microarrays were used to identify genes belonging to the σ E regulon. In total, 171 genes were found to be under the control of σ E. Of these, 101 genes had not previously been described as being σ E dependent. Disruption of some of the previously unknown genes (ydcC, yhaL, yhbH, yjaV and yqfD) resulted in a defect in sporulation.
-
-
-
Physical and gene maps of Agrobacterium biovar 2 strains and their relationship to biovar 1 chromosomes
More LessDiverse types of genomic DNA organization have been found in Rhizobiaceae, especially among Agrobacterium species. Previous studies of Agrobacterium concentrated mainly on biovar 1 strains. Little attention has been given to biovar 2 strains. The biovar 2 genome consists of a large, circular chromosome and second megabase-sized replicon, as well as several plasmids. In this study two biovar 2 strains were analysed, A. rhizogenes (A. radiobacter) K84 and A. rhizogenes A4, by constructing physical maps of their chromosomes and mega-replicons. The maps revealed that in both strains their chromosomes consist of approximately 3·7 Mbp, while the mega-replicons are 2·6 Mbp circular DNAs. Gene mapping and comparative genomic analysis were performed based on the physical maps using Southern hybridization. It was found that rDNA, as well as analysed virulence and virulence-related genes, are present only on the chromosomes. The inter-chromosomal relationship between biovar 1 and biovar 2 strains was also analysed. Interestingly, there was a high similarity between the chromosomes of biovar 2 and the circular chromosomes of biovar 1, whereas similarity among the smaller megabase-sized replicons was restricted to each biovar. Based on these observations the possible relationship among large replicons in Agrobacterium biovars 1 and 2 is discussed.
-
- Pathogens And Pathogenicity
-
-
-
Identification and characterization of a fibronectin-binding protein from Clostridium difficile
More LessA 68 kDa fibronectin-binding protein (Fbp68) from Clostridium difficile displaying significant homology to several established or putative Fbps from other bacteria was identified. The one-copy gene is highly conserved in C. difficile isolates. Fbp68 was expressed in Escherichia coli in fusion with glutathione S-transferase; the fusion protein and the native Fbp68 were purified. Immunoblot analysis and cell fractionation experiments revealed that Fbp68 is present on the surface of the bacteria. Far-immuno dot-blotting demonstrated that Fbp68 was capable of fixing fibronectin. Indirect immunofluorescence and ELISA were employed to demonstrate that C. difficile could bind both soluble and immobilized fibronectin. With competitive adherence inhibition assays it was shown that antibodies raised against Fbp68 partially inhibited attachment of C. difficile to fibronectin and Vero cells. Furthermore, Vero cells could fix purified membrane-immobilized Fbp68. Thus Fbp68 appears to be one of the several adhesins identified to date in C. difficile.
-
-
-
-
Alterations in the formation of lipopolysaccharide and membrane vesicles on the surface of Pseudomonas aeruginosa PAO1 under oxygen stress conditions
More LessIt has been postulated that phenotypic variation in the relative expression of two chemically distinct types of lipopolysaccharide (LPS), a serotype-specific LPS (B-band) and a common antigen LPS (A-band) in Pseudomonas aeruginosa is an important mechanism enabling this opportunistic pathogen to alter its surface characteristics to mediate adhesion and to survive under extreme conditions. To further investigate this, the relative expression levels of the two distinct types of LPS in P. aeruginosa PAO1 were investigated with cells grown in a chemostat at different dissolved oxygen tensions (pO2). The A-band LPS was constitutively expressed as pO2 was increased from nearly zero to 350 % of air saturation. In contrast, the B-band LPS showed a remarkable increase with increased pO2. Almost no B-band LPS was found in cells grown at a pO2 of less than 3 % of air saturation. Electron microscopic examination of cells revealed increased formation of membrane vesicles (MVs) on the surface of P. aeruginosa PAO1 under oxygen stress conditions. The toxicity of the supernatant of P. aeruginosa cultures to the growth of a hybridoma cell line significantly increased in samples taken from oxygen-stressed steady-state cultures. Furthermore, studies of adhesion in a continuous-flow biofilm culture revealed an increased adhesiveness for hydrophilic surfaces in P. aeruginosa PAO1 grown at a higher pO2. The oxygen-dependent alterations of cell-surface components and properties observed in this work provide a possible explanation for the emergence of P. aeruginosa lacking the B-band LPS in chronically infected cystic fibrosis patients. The results are also useful for understanding the processes involved in the formation of MVs in P. aeruginosa.
-
- Physiology
-
-
-
Characterization of Enterococcus faecium mutants resistant to mundticin KS, a class IIa bacteriocin
More LessThe emergence and spread of mutants resistant to bacteriocins would threaten the safety of using bacteriocins as food preservatives. To determine the physiological characteristics of resistant mutants, mutants of Enterococcus faecium resistant to mundticin KS, a class IIa bacteriocin, were isolated. Two types of mutant were found that had different sensitivities to other antimicrobial agents such as nisin (class I) and kanamycin. Both mutants were resistant to mundticin KS even in the absence of Mg2+ ions. The composition of unsaturated fatty acids in the resistant mutants was significantly increased in the presence of mundticin KS. The composition of the phospholipids in the two resistant mutants also differed from those in the wild-type strain. The putative zwitterionic amino-containing phospholipid in both mutants significantly increased, whereas amounts of phosphatidylglycerol and cardiolipin decreased. These changes in membrane structure may influence resistance of enterococci to class IIa and class I bacteriocins.
-
-
-
-
The regulatory link between carbon and nitrogen metabolism in Bacillus subtilis: regulation of the gltAB operon by the catabolite control protein CcpA
More LessBacillus subtilis assimilates ammonium by the concerted action of glutamine synthetase and glutamate synthase. The expression of the gltAB operon encoding the latter enzyme is impaired in B. subtilis ccpA mutant strains. CcpA is a pleiotropic transcriptional regulator that is the key factor in the regulation of carbon metabolism. However, in addition to their defect in catabolite repression ccpA mutants are unable to grow on minimal media with glucose and ammonium as the single sources of carbon and nitrogen, respectively. In this work, the expression of the gltAB operon was analysed and its role in growth on minimal sugar/ammonium media was studied. Expression of gltAB requires induction by glucose or other glycolytically catabolized carbon sources. In ccpA mutants, gltAB cannot be induced by glucose due to the low activity of the phosphotransferase sugar transport system in these mutants. A mutation that allowed phosphotransferase system activity in a ccpA background simultaneously restored glucose induction of gltAB and growth on glucose/ammonium medium. Moreover, artificial induction of the gltAB operon in the ccpA mutant allowed the mutant strain to grow on minimal medium with glucose and ammonium. It may be concluded that expression of the gltAB operon depends on the accumulation of glycolytic intermediates which cannot occur in the ccpA mutant. The lack of gltAB induction is the bottleneck that prevents growth of the ccpA mutant on glucose/ammonium media. The control of expression of the gltAB operon by CcpA provides a major regulatory link between carbon and amino acid metabolism.
-
- Plant-Microbe Interactions
-
-
-
Investigation of the role of a β(1–4) agarase produced by Pseudoalteromonas gracilis B9 in eliciting disease symptoms in the red alga Gracilaria gracilis
More LessGracilaria species are an important source of agar. The South African Gracilaria industry has experienced a number of setbacks over the last decade in the form of complete or partial die-offs of the agarophyte growing in Saldanha Bay, which may be attributed to bacterial infection. Since a positive correlation was observed between the presence of agarolytic epiphytes and bacterial pathogenicity, we investigated the role of an agarase in the virulence mechanism employed by a bacterium that elicits disease in Gracilaria gracilis. The recombinant plasmid pDA1, isolated from a Pseudoalteromonas gracilis B9 genomic library, was responsible for the agarolytic activity exhibited by Escherichia coli transformants when grown on solid medium. A blast search of the GenBank database showed that an 873 bp ORF (aagA) located on pDA1 had 85 % identity to the β-agarase (dagA) from Pseudoalteromonas atlantica ATCC 19262T (or IAM 12927T) at the amino acid level. AagA was purified from the extracellular medium of an E. coli transformant harbouring pDA1 by using a combination of gel filtration and ion-exchange chromatography. AagA has an M r of 30 000 on SDS-PAGE. TLC of the digestion products of AagA showed that the enzyme cleaves the β-(1,4) linkages of agarose to yield predominately neoagarotetraose. Western hybridization confirmed that the cloned agarase was in fact the extracellular β-agarase of P. gracilis B9. The observed relationship between disease symptoms of G. gracilis and the agarolytic phenotype of P. gracilis B9 was confirmed. Transmission electron microscope examination of cross sections of both healthy G. gracilis and G. gracilis infected with P. gracilis, revealed a weakening of the cell structure in the latter plants. Immunogold-labelled antibodies localized the agarase in situ to the cell walls of bleached G. gracilis. Thus, the weakening observed in the cell structure of G. gracilis infected with P. gracilis can be attributed to degradation of the mucilaginous component of the cell wall of the bleached thalli.
-
-
- Theoretical Microbiology
-
-
-
A three-dimensional, stochastic simulation of biofilm growth and transport-related factors that affect structure
More LessBiofilm structural heterogeneity affects a broad range of microbially catalysed processes. Solute transport limitation and autoinhibitor production, two factors that contribute to heterogeneous biofilm development, were investigated using BacMIST, a computer simulation model. BacMIST combines a cellular automaton algorithm for biofilm growth with Brownian diffusion for solute transport. The simulation represented the growth of microbial unit cells in a three-dimensional domain modelled after a repeating section of a constant depth film fermenter. The simulation was implemented to analyse the effects of various levels of transport limitation on a growing single-species biofilm. In a system with rapid solute diffusion, cells throughout the biofilm grew at their maximum rate, and no solute gradient was formed over the biofilm thickness. In increasingly transport-limited systems, the rapidly growing fraction of the biofilm population decreased, and was found exclusively at the biofilm–liquid interface. Trans-biofilm growth substrate gradients also deepened with increasing transport limitation. Autoinhibitory biofilm growth was simulated for various rates of microbially produced inhibitor transport. Inhibitor transport rates affected both the biofilm population dynamics and the resulting biofilm structures. The formation of networks of void spaces in slow-growing regions of the biofilm and the development of columns in the fast-growing regions suggested a possible mechanism for the microscopically observed evolution of channels in biofilms.
-
-
Volumes and issues
-
Volume 170 (2024)
-
Volume 169 (2023)
-
Volume 168 (2022)
-
Volume 167 (2021)
-
Volume 166 (2020)
-
Volume 165 (2019)
-
Volume 164 (2018)
-
Volume 163 (2017)
-
Volume 162 (2016)
-
Volume 161 (2015)
-
Volume 160 (2014)
-
Volume 159 (2013)
-
Volume 158 (2012)
-
Volume 157 (2011)
-
Volume 156 (2010)
-
Volume 155 (2009)
-
Volume 154 (2008)
-
Volume 153 (2007)
-
Volume 152 (2006)
-
Volume 151 (2005)
-
Volume 150 (2004)
-
Volume 149 (2003)
-
Volume 148 (2002)
-
Volume 147 (2001)
-
Volume 146 (2000)
-
Volume 145 (1999)
-
Volume 144 (1998)
-
Volume 143 (1997)
-
Volume 142 (1996)
-
Volume 141 (1995)
-
Volume 140 (1994)
-
Volume 139 (1993)
-
Volume 138 (1992)
-
Volume 137 (1991)
-
Volume 136 (1990)
-
Volume 135 (1989)
-
Volume 134 (1988)
-
Volume 133 (1987)
-
Volume 132 (1986)
-
Volume 131 (1985)
-
Volume 130 (1984)
-
Volume 129 (1983)
-
Volume 128 (1982)
-
Volume 127 (1981)
-
Volume 126 (1981)
-
Volume 125 (1981)
-
Volume 124 (1981)
-
Volume 123 (1981)
-
Volume 122 (1981)
-
Volume 121 (1980)
-
Volume 120 (1980)
-
Volume 119 (1980)
-
Volume 118 (1980)
-
Volume 117 (1980)
-
Volume 116 (1980)
-
Volume 115 (1979)
-
Volume 114 (1979)
-
Volume 113 (1979)
-
Volume 112 (1979)
-
Volume 111 (1979)
-
Volume 110 (1979)
-
Volume 109 (1978)
-
Volume 108 (1978)
-
Volume 107 (1978)
-
Volume 106 (1978)
-
Volume 105 (1978)
-
Volume 104 (1978)
-
Volume 103 (1977)
-
Volume 102 (1977)
-
Volume 101 (1977)
-
Volume 100 (1977)
-
Volume 99 (1977)
-
Volume 98 (1977)
-
Volume 97 (1976)
-
Volume 96 (1976)
-
Volume 95 (1976)
-
Volume 94 (1976)
-
Volume 93 (1976)
-
Volume 92 (1976)
-
Volume 91 (1975)
-
Volume 90 (1975)
-
Volume 89 (1975)
-
Volume 88 (1975)
-
Volume 87 (1975)
-
Volume 86 (1975)
-
Volume 85 (1974)
-
Volume 84 (1974)
-
Volume 83 (1974)
-
Volume 82 (1974)
-
Volume 81 (1974)
-
Volume 80 (1974)
-
Volume 79 (1973)
-
Volume 78 (1973)
-
Volume 77 (1973)
-
Volume 76 (1973)
-
Volume 75 (1973)
-
Volume 74 (1973)
-
Volume 73 (1972)
-
Volume 72 (1972)
-
Volume 71 (1972)
-
Volume 70 (1972)
-
Volume 69 (1971)
-
Volume 68 (1971)
-
Volume 67 (1971)
-
Volume 66 (1971)
-
Volume 65 (1971)
-
Volume 64 (1970)
-
Volume 63 (1970)
-
Volume 62 (1970)
-
Volume 61 (1970)
-
Volume 60 (1970)
-
Volume 59 (1969)
-
Volume 58 (1969)
-
Volume 57 (1969)
-
Volume 56 (1969)
-
Volume 55 (1969)
-
Volume 54 (1968)
-
Volume 53 (1968)
-
Volume 52 (1968)
-
Volume 51 (1968)
-
Volume 50 (1968)
-
Volume 49 (1967)
-
Volume 48 (1967)
-
Volume 47 (1967)
-
Volume 46 (1967)
-
Volume 45 (1966)
-
Volume 44 (1966)
-
Volume 43 (1966)
-
Volume 42 (1966)
-
Volume 41 (1965)
-
Volume 40 (1965)
-
Volume 39 (1965)
-
Volume 38 (1965)
-
Volume 37 (1964)
-
Volume 36 (1964)
-
Volume 35 (1964)
-
Volume 34 (1964)
-
Volume 33 (1963)
-
Volume 32 (1963)
-
Volume 31 (1963)
-
Volume 30 (1963)
-
Volume 29 (1962)
-
Volume 28 (1962)
-
Volume 27 (1962)
-
Volume 26 (1961)
-
Volume 25 (1961)
-
Volume 24 (1961)
-
Volume 23 (1960)
-
Volume 22 (1960)
-
Volume 21 (1959)
-
Volume 20 (1959)
-
Volume 19 (1958)
-
Volume 18 (1958)
-
Volume 17 (1957)
-
Volume 16 (1957)
-
Volume 15 (1956)
-
Volume 14 (1956)
-
Volume 13 (1955)
-
Volume 12 (1955)
-
Volume 11 (1954)
-
Volume 10 (1954)
-
Volume 9 (1953)
-
Volume 8 (1953)
-
Volume 7 (1952)
-
Volume 6 (1952)
-
Volume 5 (1951)
-
Volume 4 (1950)
-
Volume 3 (1949)
-
Volume 2 (1948)
-
Volume 1 (1947)
Most Read This Month
